





|
Spis treści
|
1. Droga piramidowa - przebieg
Układ piramidowy – część układu nerwowego kontrolująca ruchy dowolne i postawę ciała. Układ piramidowy posiada dwie drogi unerwiające ruchowo mięśnie. Pierwsza z nich to droga korowo-jądrowa, która unerwia mięśnie twarzoczaszki, szyi a także część mięśnia czworobocznego grzbietu. Druga to droga korowo-rdzeniowa, która unerwia resztę mięśni organizmu.
Układ piramidowy składa się z dwóch neuronów: ośrodkowego i obwodowego:
- ośrodkowy neuron ruchowy to duża, piramidowa komórka Betza, leżąca w 4 i częściowo 6 polu kory ruchowej (wg Brodmanna)
- obwodowy neuron ruchowy to komórka leżąca w rogu przednim rdzenia kręgowego lub w jądrze ruchowym nerwów czaszkowych, w zależności od tego przez jakie nerwy dany mięsień jest unerwiany.
W warstwie V kory mózgu, w okolicy tzw. zakrętu przedśrodkowego, znajduje się od 30 do 34 tysięcy komórek nerwowych piramidalnych (olbrzymich) Betza, których aksony biegną do jąder ruchowych pnia mózgu i rdzenia kręgowego. Neurony, pomiędzy którymi krążą impulsy nerwowe, są to zarówno neurony pośredniczące, znajdujące się w warstwie IV kory zakrętu przedśrodkowego, jak i neurony innych pól kory mózgu i ośrodków podkorowych. Aksony komórek Betza mają największą średnicę od 10 do 20 μm, stanowią jednak tylko około 1% włókien biegnących przez każdą drogę piramidową. Pozostałe 9% ma średnicę od 5 do 10 μm (najwięcej włókien), około 90%, ma jeszcze mniejszą średnicę od 1 do 4 μm.
Akson komórek Betza wychodząc z pola 4 lub 6 przechodzi przez istotę białą półkuli, tworząc tzw. wieniec promienisty torebki wewnętrznej (łac. corona radiata). Dalej, aksony przekazujące sygnał w kierunku mięśni zaopatrywanych przez nerwy szkieletowe), biegną przez odnogę tylną torebki (łac. crus posterior) układając się tak, że włókna związane z wyższymi partiami ciała są bardziej z przodu. Jest to tzw. droga korowo-rdzeniowa. Aksony przekazujące sygnał w kierunku mięśni unerwianych przez nerwy czaszkowe przechodzą przez kolano torebki wewnętrznej. Jest to tzw. droga korowo-jądrowa. Dalej włókna trafiają do śródmozgowia tworząc odnogi mózgu (łac. crura cerebri), gdzie włókna drogi drugiej układają się zewnętrznie w stosunku do włókien drogi pierwszej. Dalej trafiają one do mostu. Na tej wysokości włókna drogi korowo-jądrowej zaczynają się rozchodzić i kierują się do odpowiednich ruchowych jąder nerwów czaszkowych. Pozostałe trafiają do piramidy. Większość z nich krzyżuje się (przechodzi na drugą stronę rdzenia) na wysokości kaudalnej (ogonowej) części rdzenia przedłużonego wnikając do sznura bocznego. Jest to tzw. skrzyżowanie piramid, od którego dalej ciągnie się droga korowo-rdzeniowa (piramidowa) boczna. Reszta włókien tworzy drogę korowo-rdzeniową przednią. Przechodzą one dopiero na drugą stronę rdzenia na wysokości odpowiedniego neuromeru poprzez spoidło białe rdzenia kręgowego. W rogu przednim istoty szarej rdzenia kręgowego znajdują się ciała komórek obwodowych. Ich aksony opuszczają rdzeń przez korzeń przedni nerwu rdzeniowego i kierują się do mięśni efektorowych.
Skrzyżowaniu ulega 80% włókien drogi korowo-rdzeniowej, które przechodząc do sznura bocznego przeciwległej strony rdzenia kręgowego tworzą drogę korowo-rdzeniową boczną. Skrzyżowanie tej drogi wyjaśnia, dlaczego osoby praworęczne mają lepiej rozwiniętą lewą półkulę mózgu, a leworęczne prawą.
Pozostałe 20% nieskrzyżowanych włókien tworzy drogę korowo-rdzeniową przednią, biegnącą w sznurze przednim rdzenia, która oddaje stopniowo włókna do substancji szarej rdzenia kończąc się w dystalnym odcinku rdzenia kręgowego szyjnego. Droga korowo-rdzeniowa boczna kończy się na odcinku L2-L3 rdzenia kręgowego, także stopniowo oddając swoje włókna. W odcinku szyjnym ta droga oddaje aż 55% wszystkich swoich włókien, w odcinku piersiowym tylko 20%, a pozostałe 25% w odcinku lędźwiowo-krzyżowym. W rogach przednich rdzenia sygnał z pierwszego neuronu drogi piramidowej przełączany jest na neuron drugi. Twierdzenie, że drogi piramidowe są dwuneuronowe, jest uproszczeniem, ponieważ drogi dwuneuronowe stanowią zaledwie 7-15% wszystkich dróg piramidowych. W większości są to drogi wieloneuronowe. Liczba neuronów drogi zwiększa się w rdzeniu kręgowym za sprawą neuronów pośredniczących (interneuronów).
Układ piramidowy unerwia ruchowo wszystkie mięśnie poprzecznie prążkowane w całym ustroju człowieka poza jednym mięśniem, który nie jest unerwiany ruchowo przez żaden układ, mięśniem strzemiączkowym. Jest to zarazem najmniejszy mięsień poprzecznie prążkowany w organizmie człowieka i jedyny poprzecznie prążkowany, nie podlegający woli.
2. Droga piramidowa – objawy uszkodzenia
- niedowład lub porażenie,
- występowanie odruchów patologicznych (objaw Babińskiego, objaw Rossolimo),
- wzmożenie napięcia mięśni typu spastycznego (piramidowego),
- obecność wygórowanych odruchów rozciągowych typu odruch rzepkowy.
3. Obraz kliniczny w zależności od miejsca uszkodzenia drogi piramidowej
W zależności od lokalizacji uszkodzenia dróg piramidowych uszkodzenie ośrodkowego neuronu ruchowego może przyjmować następujące postacie:
- uszkodzenie kory mózgu – niedowład lub porażenie ograniczone do niewielkiej struktury np. dłoni, stopy lub jednej kończyny (tzw. monoplegia) po przeciwległej stronie od ogniska chorobowego,, której zwykle towarzyszą objawy podrażnienia kory mózgu, takie jak drgawki lub afazja,
- uszkodzenie pnia mózgu – porażenie połowicze po stronie przeciwnej od miejsca uszkodzenia (czyli porażenie połowicze lewostronne świadczy o prawostronnym uszkodzeniu pnia), zwykle towarzyszy im porażenie nerwów czaszkowych po stronie ogniska chorobowego, co daje objaw tzw. porażenia połowiczego naprzemiennego,
- uszkodzenie w obrębie torebki wewnętrznej – porażenie lub niedowład po stronie przeciwnej od ogniska chorobowego,
- uszkodzenia w obrębie rdzenia kręgowego w przypadku którego do objawów porażenia spastycznego dołączają zaburzenia pozapiramidowych dróg nerwowych, takich jak drogi przewodzące czucie, a samo porażenie jest zwykle obustronne powodując objawy tetraplegii lub paraplegii.
4. Uszkodzenie obwodowego neuronu ruchowego
Uszkodzenie ośrodkowego neuronu ruchowego spowoduje stan zwany niedowładem (łac. paresis) lub porażeniem (łac. plegia). W zależności od miejsca uszkodzenia porażenie będzie po lewej i (lub) prawej stronie ciała, przeciwstronnie (jeśli uszkodzenie znajduje się proksymalnie od skrzyżowania piramid) lub tożstronnie (jeśli znajduje się poniżej skrzyżowania). Obecne będą odruchy rozciągowe (takie jak odruch kolanowy), a ich siła będzie nawet większa, ze względu na brak sterowania mięśniem przez korę ruchową (wygórowanie odruchów ścięgnistych).
Gdy uszkodzony zostanie obwodowy neuron ruchowy, brak będzie wszelkich odruchów (nawet obronnych), mięsień będzie wiotki, ze względu na zniesienie napięcia spoczynkowego, i dojdzie do zaników mięśniowych.
Uszkodzenie górnego neuronu ruchowego:
- uszkodzenie półkuli lub pnia mózgu, mielopatia (droga korowo-rdzeniowa)
- jeśli poniżej piramid, niedowład jest po tej samej stronie co uszkodzenie
- hemipareza, parapareza
- rzadziej tetrapareza, monopareza
- wzmożenie napięcia mięśni typu spastycznego (piramidowego)
- brak lub osłabienie odruchów skórnych (brzusznych, podeszwowego)
- brak zaników mięśniowych i fascykulacji
- wygórowanie odruchów głębokich, klonusy
- obecność wygórowanych odruchów ścięgnistych
- odruchy patologiczne (Babińskiego, Rossolimo)
Uszkodzenie dolnego neuronu ruchowego:
Przyczyny:
- neuropatia ruchowa
- uszkodzenie rogów przednich rdzenia
- radikulopatia, pleksopatia, neuropatia
Objawy:
- ogniskowa męczliwość mięśni, fascykulacje
- brak lub osłabienie odruchów głebokich
- prawidłowe odruchy brzuszne i nosidłowe
- obniżone napięcie mięśniowe
- zaniki mięśniowe
- osłabione lub zniesione odruchy ścięgniste
- odruch podeszwowy (o ile nie ma niedowładu mięśni stopy)
| Uszkodzenie górnego neuronu ruchowego | Uszkodzenie dolnego neuronu ruchowego | |
| zespół rzekomoopuszkowy | zespół opuszkowy | |
| Siła mięśniowa | osłabienie mięśni / niedowład spastyczny | osłabienie pojedynczych mięśni lub grup mięśni / niedowład wiotki |
| Odruchy głębokie | wzmożone | osłabione lub nieobecne |
| Odruchy patologiczne (Babińskiego, Rossolimo) | obecne | nieobecne |
| Zaniki mięśniowe | brak zaników | obecne zaniki |
| Odruchy ścięgniste | wygórowane | osłabione lub zniesione |
| Klonusy | obecne klonusy | brak klonusów |
| Synkinezy | obecne | brak |
| Napięcie mięśniowe | spastyczność | wiotkość |
| Dyzartria | dyzartria spastyczna | dyzartria wiotka |
| Fascykulacje | brak | obecne |
5. Napięcie mięśniowe – rodzaje zaburzeń , łuk odruchowy
Napięcie mięśniowe spowodowane jest czynnością odruchów ścięgnistych. Ocenia się je, określając opór występujący przy rozciąganiu mięśnia z różną prędkością. Wzmozenie napięcia może być wywołane chorobami górnego neuronu ruchowego lub układu pozapiramidowego.
- wzmożenie napięcia typu piramidowego - spastyczność: uszkodzenie górnego neuronu ruchowego przy równoczesnym zajęciu układu siatkowatego, objaw scyzorykowy, klonusy
- sztywność pozapiramidowa, typu rury ołowianej, typu koła zębatego: w zespole parkinsonowskim i innych zespołach pozapiramidowych
- obniżone napięcie mięśniowe w dysfunkcji móżdżku następuje w wyniku zniesienia móżdżkowego torowania kory mózgu przez toniczne wyładowania jąder móżdżku
- obniżone w choreoatetozie i bazlizmie
- obniżone napięcie w uszkodzeniu dolnego neuronu ruchowego
- początkowo po uszkodzeniu górnego neuronu ruchowego pojawia się niedowład wiotki z osłabieniem lub zniesieniem odruchów ścięgnistych, zespół szlaku rdzeniowego, potem rozwija się spastyczność
6. Rodzaje uszkodzeń nerwu twarzowego i nerwu podjęzykowego
Przebieg nerwu VII:
- nerw VII utworzony jest z włókien ruchowych, zaopatrujących mięśnie mimiczne twarzy, oraz z nerwu pośredniego,prowadzącego włókna smakowe z przednich 2/3 języka i włókna przywspółczulne dla gruczołów ślinowych i mięśnia strzemiączkowego
- jądro ruchowe znajduje się w bocznej części mostu, przed opuszczeniem mostu w kącie mostowo-móżdżkowym włókna nerwu tworzą pętlę zawijającą się wokół jądra VI nerwu
- nerw wychodzi w kącie mostowo-móżdżkowym i przechodzi do przewodu słuchowego wewnętrznego razem z nerwem pośrednim i VIII nerwem czaszkowym
- wnika w głąb kości skalistej do ucha środkowego, nerw pośredni do zwoju kolanka
- nerw skalisty większy opuszcza zwój kolanka prowadząc włókna przywspółczulne dla gruczołu łzowego
- nerw VII biegnie w kanale nerwu twarzowego w części skalistej kości skroniowej, oddaje gałązkę do mięśnia strzemiączkowego, regulującą wrażliwość kosteczek słuchowych na dźwięk
- oddaje strunę bębenkową, zaopatrującą smakowo 2/3 języka i prowadzącą włókna przywspółczulne dla podżuchwowych i podjęzykowych gruczołów ślinowych
- pień nerwu VII wychodzi otworem rylcoowo-sutkowym wnika do ślinianki przyusznej i dzieli się na gałęzie zaopatrujące mięśnie mimiczne twazry
Niedowład ośrodkowy jednostronny
- spowodowany nadjądrowym uszkodzeniem GNR po stronie przeciwnej
- przyczyny: udar niedokrwienny, guz
Niedowład jednostronny obwodowy
- uszkodzenie jądra VII, zwoju kolanka lub pnia nerwu VII
- na poziomie: mostu, kąta mostowo-móżdżkowego, w kanale nerwu, zwoju kolanka, obwodowe gałęzie nerwu
Obustronny niedowład obwodowy
- obustronne porażenie Bella, borelioza, sarkoidoza, GBS, miastenia, miopatie
Uszkodzenie nerwu XII
Izolowane uszkodzenia dolnych nerwów czaszkowych są rzadkie. Przyczyny to:
- choroby neuronu ruchowego
- guzy pnia mózgu
- rak nosogardła
- guzy kłębka szyjnego
- polineuropatie, np. w przebiegu GBS
- urazy podstawy czaszki
Uszkodzenie obwodowe jednostronne:
- atrofia i fascykulacje po stronie uszkodzenia
- zbaczanie wysuniętego języka w stronę uszkodzenia
- nie ma zaburzeń połykania i artykulacji
Obustronne uszkodzenie obwodowe:
- obustronna atrofia i fascykulacje
- język porażony, na dnie jamy ustnej
- do tego dyzartria, niewielka dysfagia
- porażenie opuszkowe
Jednostronne uszkodzenie ośrodkowe:
- niewielkie zbaczanie języka w stronę przeciwną, zwykle bezobjawowe
Obustronne uszkodzenie ośrodkowe:
- ograniczenie wysuwania języka
- zespół rzekomoopuszkowy
Obustronne obwodowe uszkodzenie:
- choroba neuronu ruchowego
- jamistość opuszki
- zapalenie istoty szarej rdzenia
- obustronne procesy naczyniowe opuszki
Jednostronne obwodowe uszkodzenie
- zmiany zapalne pnia mózgu
- jamistość opuszki
- zakrzep przyśrodkowych gałęzi t. kręgowej (zespół naprzemienny Jacksona z niedowładem piramidowym przeciwstronnym)
- guz
- tętniak t. kręgowej
- rakowatość opon
- wrodzone anomalie rozwojowe okolicy foramen magnum np. zesp. Arnolda – Chiariego
- jatrogenia operacyjna
7. Zespół opuszkowy i podopuszkowy
Porażenie opuszkowe lub zespół opuszkowy – neurologiczny zespół chorobowy występujący w przypadku uszkodzenia jąder nerwów czaszkowych znajdujących się w rdzeniu przedłużonym. Dotyczy to uszkodzenia nerwów: IX, X, XI i XII na ich drodze do jąder śródmózgowia. Objawami zespołu opuszkowego są:
- zaburzenia połykania (dysfagia)
- dyzartria i niedowład podniebienia.
Najczęściej dochodzi do niego w:
- stwardnieniu zanikowym bocznym
- jamistości opuszki
- chorobach naczyniowych mózgu
- zmiany w okolicy otworu szyjnego (nowotwory pnia mózgu, guz kłębka szyjnego
- zapaleniu rogów przednich rdzenia (obecnie rzadko).
Zespół rzekomooopuszkowy (porażenie rzekomoopuszkowe) – neurologiczny zespół chorobowy powstający w wyniku obustronnego uszkodzenia dróg korowo-jądrowych biegnących do jąder nerwu językowo-gardłowego, nerwu błędnego oraz nerwu podjęzykowego. Dominującymi objawami są:
- zaburzenia połykania (dysfagia)
- dyzartria.
- Objawem odróżniającym od zespołu opuszkowego jest brak zaników mięśniowych.
Dodatkowo często stwierdza się występowanie odruchów piramidowych oraz wzmożenie odruchu żuchwowego, podniebiennego, gardłowego.
Najczęstszą przyczyną zespołu są miażdżycowe zmiany naczyniowe.
8. Korowe ośrodki mowy, rodzaje afazji
Korowe ośrodki mowy:
- Ośrodek ruchowy (ekspresywny) Broki (pole 44 i 45 Brodmana) – płat czołowy (tylna część zakrętu czołowego dolnego)
- Ośrodek czuciowy (receptywny) Wernickego (pole 22 Brodmana – płat skroniowy, pole 39 i 40 Brodmana – płat ciemieniowy – zakręt kątowy i nadbrzeżny)
- Pęczek łukowaty – strukturalna i funkcjonalna integracja obu ośrodków korowych, włókna przebiegają przez podkorową istotę białą
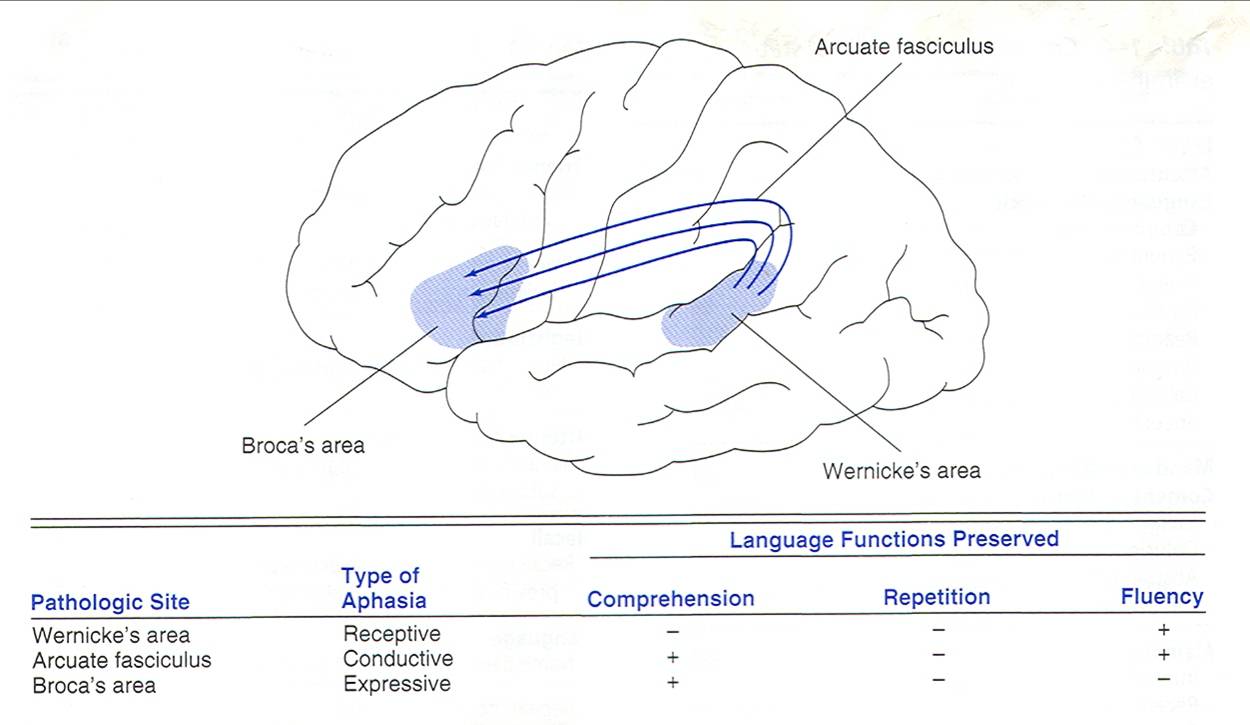
Rodzaje afazji:
- Afazja ruchowa– gdy przeważają objawy związane z zaburzeniami mówienia.
- Afazja czuciowa – gdy przeważają objawy związane z zaburzeniami rozumienia.
- Afazja mieszana (czuciowo-ruchowa) – gdy występują objawy zarówno zaburzeń mówienia, jak i rozumienia.
- Afazja całkowita – gdy występuje całkowita lub prawie całkowita niezdolność mówienia i rozumienia.
- Afazja amnestyczna – gdy główne problemy polegają na trudnościach z nazywaniem obiektów i odnajdywaniem potrzebnych słów w trakcie konwersacji (objawy te są obecne w każdej afazji, ale mogą występować także oddzielnie, najczęściej jako stan zejściowy po poważniejszych zaburzeniach afatycznych).
9. Przyczyny dyzartrycznych zaburzeń mowy
Dyzartia - termin z zakresu neurologii; jeden z typów zaburzeń mowy, wynikający z dysfunkcji aparatu wykonawczego (języka, podniebienia, gardła, krtani). Dysfunkcja może być spowodowana: uszkodzeniem mięśni, unerwiających ich nerwów czaszkowych, jąder tych nerwów, dróg korowo-jądrowych, układu pozapiramidowego.
- Dyzartria spastyczna (drogi korowo-jądrowe – zespół rzekomoopuszkowy)
- Dyzartria pozapiramidowa (hipokinetyczna–parkinsonizm, hiperkinetyczna- pląsawica, dystoniczna)
- Dyzartria ataktyczna (móżdżkowa)
- Dyzartria wiotka (zespół opuszkowy – uszkodzenie jąder lub pni nn X, XII, (VII), miastenia, miopatia)
10. Budowa układu pozapiramidowego
Układ pozapiramidowy (układ podkorowy, układ ruchowy prążkowiowy) wraz z układem piramidowym bierze udział w wykonywaniu przez organizm czynności ruchowej. Jeśli jednak układ piramidowy zajmuje się czynnościami, które wymagają od nas skupienia (np. nauka, jazdy na rowerze, nauka pisania), to układ pozapiramidowy powoli przejmuje i automatyzuje czynności, które wcześniej były pod kontrolą układu piramidowego. Układ pozapiramidowy jest więc układem wspomagającym, odciążającym nas od skupiania się nad codziennymi czynnościami, umożliwiający nam pewną automatyzację. Współdziała w wyzwalaniu ruchów dowolnych i regulowaniu napięcia mięśni poprzecznie prążkowanych.
W układzie pozapiramidowym wyróżnia się następujące składowe anatomiczne:
- prążkowie (jądro ogoniaste + skorupa)
- gałkę bladą
- jądro niskowzgórzowe
- jądra wzgórza: brzuszne przednie, brzuszne boczne i środkowo-pośrodkowe
- istotę czarną
- jądro konarowo-mostowe
- połączenia tych struktur
Część tylna zakrętu czołowego górnego - złożone ruchy tułowia
Część tylna zakrętu czołowego środkowego - koordynacja ruchów głowy i gałek ocznych
Część tylna zakrętu czołowego dolnego - dźwięki artykułowane
Ciało prążkowane - ruchy zautomatyzowane, napięcie mięśniowe
Jądro niskowzgórzowe - różnicowanie jakości impulsów ich umiejscowienia, balansowanie kończyn
Istota czarna - koordynacja ruchów mimowolnych, ruchy szybkie
Jądro czerwienne - koordynacja czynności ośrodków układu z czynnością kory mózgu, móżdżku, jąder przedsionka
11. Neurotransmitery układu pozapiramidowego
Pobudzające: ACh, dopamina, glutaminian
Hamujące: GABA, peptydy opioidowe
W istocie czarnej dopamina
Prążkowie-gałka blada GABA
We wzgórzu glutaminian
W korze glutaminian
W prążkowiu ACh
Konsekwencją zaburzenia funkcji neuronów istoty czarnej w parkinsonizmie jest niedobór dopaminy (ok. 70-80%) w istocie czarnej i prążkowiu, i przewaga aktywności neuronów glutaminergicznych, hamujących jądra wzgórza. Stąd w leczeniu stosuje się L-DOPĘ, amantadynę zwiększajaca uwalnianie endogennej dopaminy, agonistów receptorów D (np. bromokryptyna), inhibitory MAO (selegilina), inhibitory COMT (tolkapon, entakapon), leki antycholinergiczne (triheksfenidyl).
W chorobie Alzheimera obserwuje się nadmierne pobudzenie neuronów glutaminergicznych, dlatego hamuje się je stosując inhibitory NMDA np. memantynę. Ponadto stosuje się leki hamujące acetylocholinesterazę (rywastygminę, donepezil, galantaminę).

12. Najważniejsze rodzaje klinicznych zespołów pozapiramidowych
- Zespół parkinsonowski – zespół hipertoniczno-hypokinetyczny (zanik komórek istoty czarnej, i jader zawierających melaninę w gałce bladej i ciele prążkowanym)
- Pląsawica – zespół hypotoniczno-hyperkinetyczny (zanik małych komórek w jądrze ogonisatym i skorupie)
- Atetoza - ruchy mimowolne, arytmiczne o zmiennym umiejscowieniu, powolne, zaakcentowane dystalnie, ekstremalne wygięcie w stawach ( skorupa, jadro ogoniaste, zewnętrzna część gałki bladej)
- Balizm i hemibalizm - mimowolne, nierytmiczne, błyskawiczne, obszerne ruchy wielu odcinków kończyn równocześnie (jadro niskowzgórzowe Luysa)
- Dystonia - skurcze pojedynczych mięśni lub grup mięśniowych ( skorupa, wzgórze)
- Mioklonie - mimowolne, nieregularne, szybkie krótkotrwałe drgnięcie pojedynczych mięśni lub grup mięśniowych (jadro zębate, dolna część oliwki, jądro czerwienne, ewentualnie obwodowy układ nerwowy
13. Rodzaje ruchów mimowolnych
- Drżenie
- Pląsawica
- Hemibalizm
- Dystonia
- Atetoza
- Mioklonie
14. Przykłady nabytych zaburzeń pozapiramidowych (w chorobach ogólnoustrojowych, polekowe, po zatruciach)
- parkinsonizm po śpiączkowym zapaleniu mózgu
- parkinsonizm po zanieczyszczonym narkotyku (MPTP)
- parkinsonizm po zatruciu CO, H2S, manganem, talem, ołowiem
- chorea minor w przebiegu gorączki reumatycznej (pląsawica Sydenhama)
- ruchy choreoatetotyczne w dyskinezach późnych u pacjentów leczonych lewodopą, pochodnymi feotiazyny lub butyrofenonu
- choreoatetoza w czerwienicy prawdziwej
- mioklonie po anoksji
- asteriksje w encefalopatii wątrobowej lub mocznicowej
15. Fizjologiczna rola układu pozapiramidowego
- Rozpoczęcie i wykonywanie czynności ruchowej somatycznej
- Wpływa na automatyczną stereotypową aktywność ruchową związana z utrzymaniem postawy oraz odruchami
- Wpływa na czynność ruchowa poprzez wzgórze, korę mózgową, układ korowo-opuszkowy i krorowo-rdzeniowy
16. Kliniczne objawy uszkodzenia móżdżku
Zespół móżdżkowy – neurologiczny zespół chorobowy występujący w uszkodzeniu móżdżku, objawiający się zaburzeniami chodu oraz niezbornością ruchową (ataksja). Dodatkowo często występuje skandowana mowa, drżenie zamiarowe, oczopląs, obniżone napięcie mięśniowe.
Etiologia
- guzy móżdżku: przerzutowe lub pierwotne (oponiak, schwannoma nerwu VIII, medulloblastoma, haemangioblastoma), zespół paraneoplastyczny
- stwardnienie rozsiane
- udar krwotoczny
- ropień móżdżku
- wirusowe zapalenie mózgu
- zatrucie lekami przeciwpadaczkowymi
- ostre zatrucie alkoholem
- cerebellopatia alkoholowa związana z przewlekłym nadużywaniem alkoholu
- niedoczynność tarczycy
- uraz głowy
- wady wrodzone:
- malformacja Arnolda-Chiariego
- wrodzone zwężenie wodociągu mózgu
- malformacja Dandy'ego-Walkera
- wrodzone ataksje móżdżkowe:
- ataksja Friedreicha
- dziedziczne ataksje rdzeniowo-móżdżkowe
Zespół robaka przedniego
- Zmiany patologiczne: obszar płata przedniego (kończyna dolna)
- Zanik górnej części robaka
- Ataksja chodu, tułowia, kończyn dolnych
- Przyczyna: nadużywanie alkoholu
Zespół robaka tylnego
- Zmiany patologiczne: płat grudkowo-kłaczkowy
- Ataksja tułowia i chodu, kończyn górnych i dolnych
- Przyczyny: guzy (rdzeniaki i wyściółczaki)
Zespół półkulowy (pancerebellarny)
- Zmiany patologiczne: półkula móżdżku
- Ataksja : kończyn górnych i dolnych, tułowia i chodu po stronie uszkodzenia
- Przyczyna: ropień lub guz móżdżku
Zatrucie fenytoiną
- Ataksja, oczopląs, zaburzenia chodu, mowy
17. Przebieg drogi rdzeniowo-wzgórzowej i drogi czucia głębokiego
- Droga trzyneuronowa
- Neurony I-rzędowe w zwojach korzeni tylnych
- Neurony II-rzędowe w istocie szarej rdzenia ipsilateralnie
- Aksony neuronów II-rzędowych przechodzą na drugą stronę i kończą się w kontralateralnym wzgórzu
- Neurony III-rzędowe we wzgórzu, projekcja do kory somatoczuciowej
- Organizacja somatotopowa (znana mapa części ciała)
- Transmisja synaptyczna (I→II→III neuron) może być modulowana przez inne neurony (inhibicja lub potencjacja)
Droga rdzeniowo-wzgórzowa
Zwoje korzeni tylnych (I neuron) → rogi tylne rdzenia (II neuron) → aksony przechodzą na drugą stronę przez spoidło przednie na wszystkich poziomach segmentalnych → kierują się ku górze w przednio-bocznej części rdzenia jako szlak rdzeniowo-wzgórzowy boczny (Lissauera) (w sznurach bocznych – czucie bólu i temperatury) i szlak rdzeniowo-wzgórzowy przedni (w sznurach przednich – czucie dotyku) → oba szlaki zbiegają się w pniu mózgu jako wstęga rdzeniowa, sąsiadująca z wstęgą przyśrodkową → brzuszne tylne jądro wzgórza (III neuron) → projekcja do kory somatoczuciowej
Funkcja
- czucie dotyku bez określenia jego rodzaju
- czucie bólu
- czucie ciepła
- czucie zimna
Uszkodzenie
- zniesienie czucia bólu i temperatury
- osłabienie czucia dotyku
- poziom zaburzeń 2-3 segmenty poniżej poziomu uszkodzenia (ukośny przebieg włókien przez spoidło przednie)
Objawy uszkodzenia:
- parestezje
- ból
- narażenie na urazy
18. Rozszczepienie zaburzenia czucia – definicja, przyczyny, lokalizacja uszkodzenia
Rozszczepienne zaburzenia czucia - przebiegające z zaoszczędzeniem czucia dotyku, czucia wibracji i czucia prpprioceptywnego.
Zmiany dolnej części mostu, rdzenia przedłużonego i górnej części rdzenia kręgowego mogą powodować rozszczepienne zaburzenia czucia bólu i temperatury w obrębie twarzy.
Albo utrata czucia rdzeniowo-wzgórzowego przy zachowanej czynności sznurów tylnych w uszkodzeniu środkowej części rdzenia.
Rozszczepienny charakter zaburzeń czucia między modalnościami bólu i temperatury a modalnością rodzaju dotyku, rozróżniania dwóch dotykanych punktów, wibracji i propriocepcji występuje w:
- uszkodzeniu środkowej części rdzenia, np. przez demielinizację
- połowiczym uszkodzeniu rdzenia
- uszkodzeniu bocznej części rdzenia przedłużonego
- niedoborze witaminy B12
- uszkodzeniu przedniej części rdzenia.
Zespół wewnątrzrdzeniowy – uszkodzenie środkowej części rdzenia. Obustronnie osłabione czucie bólu i temperatury, zachowane czucie dotyku, + objawy zespołu GNR + objawy zespołu DNR + zespół tylnosznurowy.
19. Najczęstsze rodzaje zaburzeń chodu w neurologii
Chód nieprawidłowy może być symetryczny lub asymetryczny.
- Chód hemiparetyczny: zgięcie w stawie łokciowym i wewnętrzna rotacja kończyny górnej po stronie niedowładu (postawa Wernickego-Manna)
- Chód parkinsonowski: bez balansowania kończynami górnymi, zgarbiony
- Chód w ataksji móżdżkowej: na szerokiej podstawie, zmienny, chód pijanego
- Chód ataktyczny tylnosznurowy: chód chwiejny, na szerokiej podstawie, często z przytupywaniem
- Chód brodzący (steppage)
- Chód w rwie kulszowej
- Chód perypatetyczno-spastyczny
- Chód kurczowo-bezładny
- Chód miopatyczny (kaczkowaty) – dystrofie, niedowład proksymalnych mięśni kończyn dolnych
- Apraksja chodu - chód niepewny, jak po lodzie
- Chód histeryczny - zaburzenia konwersyjne, chód dziwaczny i zmienny
Prawidłowy chód wymaga współdziałania układów
- piramidowego
- pozapiramidowego
- móżdżkowego
- obwodowego neuronu ruchowego (ruchowe nerwy czaszkowe, nerwy rdzeniowe)
Uwzględnia informację czuciową proprioceptywną, wzrokową i przedsionkową o ułożeniu i ruchu ciała
Zaburzenia ruchowe – uszkodzenie jednego lub kilku układów
20. Różnicowanie ataksji móżdżkowej i tylnosznurowej
Chód tylnosznurowy – zamknięcie oczu powoduje nasilenie zaburzeń chodu; próba Romberga (+)
Chód móżdżkowy – tendencja do padania w stronę uszkodzenia w zespole półkulowym
21. Agnozja – rodzaje, obraz kliniczny
Agnozja - to zaburzenie odbioru bodźców wzrokowych, słuchowych lub czuciowych na skutek uszkodzenia tkanek mózgu. Jest to niemożność rozpoznania znajomych przedmiotów pomimo prawidłowej percepcji wzrokowej, słuchowej lub dotykowej, a także prawidłowej pamięci, języka i ogólnej sprawności intelektualnej
Z uwagi na różne rodzaje modalności wyróżnia się:
- agnozję wzrokową
- agnozję słuchową
- agnozję dotykową
Agnozja występuje w uszkodzeniach kory mózgowej, szczególnie II- rzędowych i III-rzędowych obszarów analizatora danej modalności zmysłowej (kojarzeniowej kory wzrokowej, słuchowej lub somatosensorycznej, gdzie odbywa się integracja informacji sensorycznej różnego typu).
22. Apraksja – definicja, rodzaje, obraz kliniczny
Apraksja - trudność egzekwowania wyuczonych czynności ruchowych, która nie wynika z niedowładu, niezborności ani zaburzeń czucia, jak również trudności rozumienia ze strony chorego.
- uszkodzenie lewego płata ciemieniowego z przerwaniem połączeń kojarzeniowych między stykiem skroniowo-ciemieniowym i korą przedruchową w płacie czołowym - apraksja wyobrażeniowa (ideacyjna) obejmująca obie strony ciała
- uszkodzenie kory przedruchowej (pole 6 Brodmana, szczególnie część przyśrodkowa, tzw. dodatkowe pole ruchowe – planowanie i programowanie złożonych czynności ruchowych) - apraksja ruchowa (motoryczna)
- uszkodzenie włókien spoidłowych pomiędzy obszarami ruchowymi lewego i prawego płata czołowego - apraksja tylko w lewych kończynach

23. Obraz zaburzeń czuciowych w zespole Brown-Sequarda: całkowitym, poprzecznym uszkodzeniu rdzenia kręgowego
Zespół Browna-Séquarda to zespół neurologiczny spowodowany jednostronnym, poprzecznym uszkodzeniu połowy rdzenia kręgowego.
Na obraz kliniczny zespołu w czystej postaci składają się:
- tożstronny niedowład piramidowy
- tożstronne zaburzenia czucia wibracji i czucia głębokiego
- przeciwstronne zaburzenia czucia bólu i temperatury.
24. Obraz zaburzeń czucia w uszkodzeniach opuszki i polineuropatiach
Zespół Wallenberga (zespół boczny opuszki) – zespół neurologiczny spowodowany zamknięciem tętnicy móżdżkowej tylnej dolnej (PICA). Zespół przebiega z:
- zespołem Hornera
- porażeniem nerwów V, IX i X
- niedosłuchem
- oczopląsem
- niezbornością
- drżeniem zamiarowym po stronie uszkodzenia, a także:
- zaburzeniami czucia ciepła i bólu na twarzy
- porażeniem zwieraczy gardła
- zaburzeniami czucia bólu i ciepła na kończynach i tułowiu
- zaburzeniami mowy i zawrotami głowy po stronie przeciwnej.
25. Unaczynienie tętnicze mózgu
Dopływ krwi do mózgu zapewniają 4 tętnice: 2 tętnice szyjne wewnętrzne i 2 kręgowe. Tętnice kręgowe łącząc się łącząc się tworzą tętnicę podstawną, która dzieli się na 2 tętnice tylne mózgu. Każda z tętnic tylnych łączy się z tętnicą szyjną wewn. za pośrednictwem tętnicy łączącej tylnej. Po jednej i po drugiej stronie od tętnicy szyjnej wewn. odchodzi tętnica mózgu przednia, tętnice mózgu przednie łączy tętnica łącząca przednia. Koło tętnicze Willisa tworzy najważniejsze, ale nie jedyne połączenie między tętnicami mózgu. Krążenie oboczne może rozwinąć się też za pośrednictwem połączeń między tętnicą szyjną zewn. i wewn.: połączenie pomiędzy końcowymi odgałęzieniami tętnicy szczękowej zewnętrznej a tętnicą oczną. Korowe gałęzie tętnic mózgu tworzą połączenia zwane oponowymi.
26. Udar mózgu – definicja, rodzaje
Ogniskowe lub globalne zaburzenia czynności mózgu o etiologii naczyniowej, występujące nagle i utrzymujące się dłużej niż 24 godz. Światowa Organizacja Zdrowia określa mianem udaru mózgu.
Udar mózgu stanowi trzecią co do częstości występowania przyczynę zgonów na świecie, w Polsce czwartą z powodu dużej ilości urazów komunikacyjnych.
w zależności od mechanizmu patogenetycznego
- zakrzepowo–zatorowe
- zatorowe
- zatokowe (lakunarne)
- hemodynamiczne, związane z zaburzeniami perfuzji w oun, wynikającej z nagłej, uogólnionej niedostateczności krążenia.
w zależności od dorzecza naczyniowego objętego niedokrwieniem
- zawał mózgu w obszarze całego przedniego unaczynienia
- częściowy zawał mózgu z zakresu przedniego unaczynienia
- zawał zatokowy
- zawał mózgu w obszarze unaczynienia tylnego
w zależności od dynamiki objawów
- przemijający napad niedokrwienny mózgu (TIA)
- odwracalny, niedokrwienny ubytek neurologiczny (RIND)
- zawał mózgu (udar mózgu dokonany)
27. Niemiażdżycowe przyczyny zakrzepów w tętnicach mózgowych
90% - miażdżyca
Zatory najczęściej są spowodowane skrzeplinami pochodzącymi z serca lub dużych naczyń (zator naczynie-naczynie).
Zatory naczyń mózgowych
- pochodzenia sercowego:
- zapalenie wsierdzia
- wady zastawkowe
- zawał serca
- migotanie przedsionków.
Pochodzenia pozasercowego:
- skrzepliny przyścienne z dużych tętnic
- skrzepliny z układu żylnego w przypadku otworu w przegrodzie serca.
Zatory tłuszczowe, gazowe, nowotworowe
28. Udar na tle zatokowym: przyczyny, obraz kliniczny
- zwykle u osób z nieleczonym przewlekłym nadciśnieniem
- około 25% udarów niedokrwiennych
- Zazwyczaj jako skutek choroby małych naczyń przeszywających od tętnicy środkowej mózgu
- Obejmuje struktury podkorowe: jądra podstawy, wzgórze, pień mózgu, torebkę wewnętrzną
- lipohialinoza drobnych naczyń przeszywających od tętnicy naczyniówkowej przedniej (prążkowiowo-torebkowej) lub zmiany zakrzepowo-zatorowe arterioli
- powstają zawały podkorowe (lakuny) o średnicy do 15 mm i małe tętniaki Charcota-Boucharda
29. Udar hemodynamiczny
Zaburzenia perfuzji w obrębie oun, wynikające z nagłej uogólnionej niedostateczności krążenia.
Zaburzenia hemodynamiczne wywołane są spadkiem systemowego ciśnienia tętniczego krwi o różnej etiologii, przekraczającym możliwości autoregulacyjne krążenia mózgowego.
Spadki pojemności minutowej serca w przebiegu:
chorób mięśnia sercowego, zaburzeń rytmu, w zatrzymaniu krążenia, spowodowane lekami hipotensyjnymi, diuretykami.
30. Czynniki ryzyka udarów niedokrwiennych mózgu
Niemodyfikowalne:
- wiek – ryzyko zwiększa się 2-krotnie co 10 lat
- płeć męska
- etniczne (rasa czarna i żółta)
- predyspozycje rodzinne i genetyczne (udar w rodzinie w wywiadzie, genetycznie uwarunkowane zespoły predysponujące do stanów zakrzepowych, hiperhomocysteinemia)
Modyfikowalne:
- nadciśnienie tętnicze
- choroby serca (migotanie przedsionków)
- cukrzyca
- palenie papierosów
- nadużywanie alkoholu
- otyłość
- hipercholesterolemia i dyslipidemia
- zaburzenia krzepnięcia, w tym polekowe
- przebyty udar bądź przemijający atak niedokrwienny (TIA)
31. Profilaktyka wtórna udarów niedokrwiennych na tle zatorowym i niezatorowym
- zachęcanie do rzucenia palenia
- leczenie zaburzeń lipidowych
- kontrola glikemii
- podawanie ASA w dawce 150 mg/d albo klopidogrelu
- leczenie przeciwkrzepliwe
- antybiotyki w zapaleniu wsierdzia
- leki antyarytmiczne w migotaniu przedionków
- odstawienie leków prozakrzepowych
- kontrola ciśnienia tętniczego
32. Podział dynamiczny udarów
w zależności od dynamiki objawów
- przemijający napad niedokrwienny mózgu (TIA) - ogniskowe objawy neurologiczne, do 24 h
- odwracalny, niedokrwienny ubytek neurologiczny (RIND) - objawy neurologiczne ustępują w ciągu 3 tygodni
- zawał mózgu (udar mózgu dokonany) - objawy utrzymujące się ponad 3 tygodnie
33. Postępowanie w ostrej fazie udaru niedokrwiennego
Postępowanie w ostrym okresie udaru niedokrwiennego:
Szybkie zdiagnozowanie, szybka hospitalizacja (Oddział Udarowy), szybko podjęta terapia farmakologiczna, wczesna rehabilitacja, szybkie podjecie prewencji wtórnej.
- Leczenie antyagregacyjne
- Leczenie trombolityczne (wczesna rekanalizacja za pomocą rekombinowanego tkankowego aktywatora plazminogenu, w ciagu 3 h)
- Leczenie przeciwzakrzepowe
Leczenie antyagregacyjne: zastosowanie w okresie 48 h od wystąpienia udaru kwasu acetylosalicylowego (300 i 160 mg)
Leczenie trombolityczne: rekombinowany tkankowy aktywator plazminogenu, który aktywuje przejście plazminogenu w plazminę. Leczenie należy wdrożyć w ciągu pierwszych 3 h.
Leczenie przeciwzakrzepowe: heparyna i jej drobnocząsteczkowe pochodne (Fraxiparyna i Calciparyna) – narastanie zakrzepu w układzie kręgowo- podstawnym, zatorowej przyczyny udaru, powtarzających się incydentów TIA.
34. Przyczyny krwotoków śródmózgowych
35. Krwawienie podpajęczynówkowe: przyczyny, objawy
Przyczyny i czynniki ryzyka
- tętniaki workowate 70%
- malformacje tętniczo-żylne 10%
- inne 20%
Ryzyko krwotoku podpajęczynówkowego jest większe u ludzi z nadciśnieniem tętniczym, miażdżycą i chorobami prowadzącymi do zaburzeń krzepnięcia krwi, takich jak białaczka.
Objawy
Do objawów krwotoku podpajęczynówkowego należą:
- nagły, ostry i bardzo silny ból głowy, który osiąga maksymalne nasilenie w ciągu kilku sekund (pacjenci czasem opisują ten ból jako "uderzenie młotkiem w głowę" lub najsilniejszy ból, jaki kiedykolwiek mieli w życiu - również w porównaniu z bólem porodowym);
- zawroty głowy, mdłości, wymioty i drgawki
- zaburzenia częstości oddechu i tętna
- utrata przytomności
- sztywność karku
- światłowstręt
- inne zaburzenia i objawy neurologiczne (na przykład drętwienie albo paraliż jednej kończyny, nierówna szerokość źrenic, upośledzenie mowy).
Do wstępnej oceny stanu klinicznego chorego z krwotokiem podpajęczynówkowym używana jest skala Boterella w modyfikacji Hunta i Hessa:
- I° – Lekki ból głowy, zaznaczona sztywność karku
- II° – Średni lub silny ból głowy, wyraźne objawy oponowe (duża sztywność karku), objawy uszkodzenia nerwów czaszkowych (głównie gałkoruchowych)
- III° – Nieduże zaburzenia przytomności (senność) i (lub) jakościowe świadomości, obecne objawy ogniskowe
- IV° – Znaczne zaburzenia przytomności (sopor), niedowład połowiczy, zakłócenia czynności wegetatywnych (objawy odkorowania)
- V° – Głęboka śpiączka, sztywność odmóżdżeniowa, prężenia tylnojamowe.
36. Krwawienie podpajęczynówkowe: postępowanie diagnostyczne, terapeutyczne
Diagnostyka:
- CT
- MRI
- PMR
- MRA
- Angiografia/DSA
Leczenie:
- Jak najszybciej chirurgiczne
- Operacja zabezpieczenia krwiaka
- Zwalczanie obrzęku mózgu
- Kontrola internistyczna
- Zwalczanie kaszlu, zaparć
- Postępowanie zachowawcze
37. Krwawienie podpajęczynówkowe: powikłania wczesne i późne
Wczesne:
- skurcz naczyń
- obrzęk mózgu, cytotoksyczny lub naczyniowy
- śmierć
- napady padaczkowe
- zaburzenia układu krążenia
- neurogenny obrzęk płuc
- zaburzenia wodno-elektrolitowe
Późne:
- obrzęk mózgu
- niedożywienie
- depresja
- odleżyny
- zakrzepica żył głębokich, zatorowość płucna z uieruchomienia
- napady padaczkowe
- wodogłowie normotensyjne
38. Stwardnienie rozsiane – patomorfologia
Stwardnienie rozsiane (SM, sclerosis multiplex) jest przewlekłą chorobą, która zaczyna się najczęściej u młodych dorosłych. Charakteryzuje się w obrazie neuropatologicznym licznymi ogniskami zapalenia, demielinizacji oraz bliznami glejowymi rozmieszczonymi w istocie białej ośrodkowego układu nerwowego.
39. SM – przebieg i rokowanie
Postacie SM:
- zespół izolowany klinicznie (CIS)
- nawracajaco-remitująca (relapsing-remitting, RRSM) 85-90%
- wtórnie postępująca (secondary progressive, SPSM) u przynajmniej 1/2 z RRSM
- pierwotna postępująca (primary progressive, PPSM) - 10%
- postępująco-nawrotowa (progressive-relapsing, PRSM) - najrzadsza
Przebieg kliniczny choroby jest różnorodny: od stanu łagodnego do schorzenia szybko postępującego i prowadzącego do inwalidztwa.
40. SM – objawy kliniczne
SM charakteryzuje się tym, że zmiany są rozsiane w czasie i przestrzeni.
Do najczęstszych objawów należą:
- Ruchowe- niedowłady spastyczne
- Czuciowe- zaburzenia czucia, bóle, objaw Lhermitte’a, objaw Uhthoffa
- Móżdżkowe- ataksja, drżenia, oczopląs, dyzartria
- Nerwy czaszkowe ( pień mózgu )- zaburzenia wzroku, zaburzenia gałkoruchowe, nerwy czaszkowe: V, VII, VIII, objawy opuszkowe, zawroty głowy
- Autonomiczne- zaburzenia zwieraczy, zaburzenia czynności seksualnych, pocenie się, zaburzenia naczynioruchowe
- Objawy psychiatryczne- depresja, euforia, zaburzenia funkcji poznawczych
- Objawy wzrokowe obejmują dwojenie, wrażenie niewyraźnego widzenia, osłabienie ostrości wzroku lub jego utrata po jednej albo obu stronach, zaburzenie pola widzenia od jednostronnego mroczka lub ograniczenia pola widzenia do niedowidzenia połowiczego.
Typowo objawy te rozpoczynają się w ciągu kilku godzin lub dni. Pacjenci mogą mieć problemy w rozpoznawaniu przedmiotów lub twarzy, często określane jako wrażenie nieostrego widzenia
We wczesnych stadiach pozagałkowego zapalenia nerwu wzrokowego lub w zapaleniu o łagodnym przebiegu mogą pojawić się zaburzenia widzenia barw, podczas, gdy widzenie czarno-białe pozostaje prawidłowe.
Objawy rzadziej obserwowane w SM- napady padaczkowe, bóle głowy, nerwoból nerwu trójdzielnego.
41. SM – metody diagnostyczne
Badania dodatkowe:
- Rezonans magnetyczny (MRI) – w 90% przypadków z SM wykazuje liczne ogniska w istocie białej i stanowi badanie z wyboru. Ocenia się zmiany w obrazach T2- zależnych albo w sekwencji FLAIR
- Badanie MRI u 90% chorych wykazuje liczne ogniska w istocie białej i stanowi badanie z wyboru w diagnostyce SM.
- Badanie płynu mózgowo–rdzeniowego: Charakterystyczne zmiany gammaglobulin (IgG). Wykrywane są w elektroforezie płynu jako prążki oligoklonalne i stwierdzane u 90% pacjentów z klinicznie pewnym SM.
- Korowe potencjały wywołane: wzrokowe, słuchowe i somatyczne: wydłużone latencje potencjałów
42. SM – co to jest rzut w SM, postępowanie farmakologiczne
Rzutem nazywa się wystąpienie nowego objawu lub nasilenie już istniejącego, trwające 24 h i więcej i powodujące pogorszenie stanu chorego o 1 i więcej pkt w skali EDSS (Rozszerzona Skala Niesprawności Ruchowej).
Leczenie rzutu choroby:
- Kortykosterydy
- Methylprednisolon (Solumedrol) dożylnie w dawce 1g/d. przez 5 dni
- Na zakończenie kuracji można podać prednison doustnie w zmniejszających się dawkach: 80 mg dz. przez 4 dni, następnie 60, 40, 20, 10 i 5 po 4 dni.
43. Leczenie objawowe w SM
Leczenie objawowe:
Obejmuje zwalczanie objawów składających się na obraz kliniczny SM: spastyczność, zaburzenia zwieraczy, niedowłady, niezborność, drżenie, zawroty głowy, dysfunkcja seksualna, zespół zmęczenia, parestezje i inne.
Spastyczność:
- Baclofen
- Sirdalud
- toksyna otulinowa
Nietrzymanie moczu:
- Ballapan
- Ditropan
Zatrzymanie moczu:
- Prostygmina
- Pilokarpina
- Guanetydyna
Bóle napadowe:
- Karbamazepina
- Gabapentyna
Bóle przewlekłe:
- niesterydowe leki przeciwzapalne
- amitryptylina.
44. Leczenie SM mające na celu spowolnienie postępu choroby
Leczenie modyfikujące przebieg choroby:
- Interferon beta 1A (Avonex)
- Interferon beta 1A (Rebif)
- Interferon beta 1B (Betaseron)
- Octan glatirameru (Copaxon lub Copolymer I)
- Mitoksantron (Novantrone)
- Natalizumab (Tisabri)
45. Samoistne bóle głowy, etiopatogeneza, podział, objawy kliniczne
Bóle samoistne
- Ból głowy mediatorowy-wskutek uwalniania substancji (mediatorów) bólotwórczych: migrena, klasterowy ból głowy
- Migrena (migrena)
- Ból głowy typu napięciowego (napięciowy ból głowy)
- Ból głowy klasterowy (klasterowy ból głowy)
- Przewlekła napadowa hemikrania (przewlekła napadowa hemikrania)
- Nerwobóle (np. neuralgia nerwu V)
Bóle głowy samoistne
- Migrena
- Napięciowy ból głowy
- Klasterowy ból głowy i przewlekła napadowa hemikrania
- Przewlekły klujący ból głowy
- Ból głowy po zjedzeniu lodów
- Kaszlowy i wysiłkowy ból głowy
- Ból głowy związany z aktywnością seksualną
- Karotydynia
Postacie migreny:
- Migrena bez aury
- Migrena z aurą (wzrokowa, hemiparetyczna, skojarzona, afatyczna)
- Migrena podstawna (omdleniowa)
- Aura migrenowa bez bólu głowy
- Migrena okoporażna
- Migrena siatkówkowa
- Dziecięce zespoły migrenowe (ekwiwalenty migreny)
- Powikłania migreny (stan migrenowy, migrenowy zawał mózgu)
Klasterowy ból głowy:
- Częstość: 0,05-0,1%, 13-70 razy wyższa wśród członków rodziny
- M>K (4:1 do 7:1) 20-30 r.ż.
- Silny jednostronny ból okolicy oczodołu, nadoczodołowej, skroniowej od 15 do 180 min. od 1-2 dni do 8/dobę, budzi ze snu
- Co najmniej jeden z wymienionych objawów po stronie bólu: przekrwienie spojówek, łzawienie, zatkanie nosa, wyciek z nosa, pocenie czoła lub twarzy, zwężenie źrenicy, opadnięcie powieki, obrzęk powieki.
- Konieczne spełnienie jednego z warunków, wykluczenie innych bólów głowy, innych przyczyn, w razie istnienia innej choroby brak z nią związku.
- Mogą być tzw. klastery (14 dni do roku)
Zwalczanie pojedynczego napadu:
- Wdychanie 02 7l/min przez 15min.
- Sumatriptan 6-12mg s.c.
Przerywanie klasteru:
- Steroidy: dexametason i.v. 16mg/d
Leczenie profilaktyczne:
- Kwas walproinowy 600-1800mg/d
- werapamil 320-480mg/d
- węglan litu 100mg/d
Przewlekła napadowa hemikrania (CPH)
- Częstość 0.0005-0.002%
- K>M (3-5:1) 20-40r.ż
- Połowicze bóle głowy z promieniowaniem do karku i ramienia
- Czas trwania:2-45 min. Śr 15 min, 5-40 d (śr.13/d)
- Przebieg przewlekły
- Poprawa po indometacynie (150 mg doustnie)
Napięciowy ból głowy:
- Częstość: 20-40%, Czas trwania bólu: od 30min. do 7 dni.
- K>M 2:1 do 4:1
- Charakter bólu: uciskowy, opasujący, nie pulsujący, łagodna lub średnia intensywność, okolica czołowa, ciemieniowo- potyliczna, obustronnie, brak nasilenia pod wpływem aktywności fizycznej.
- Brak obu objawów: nudności i wymioty, foto- i fonofobia (może być jeden).
- Brak innej choroby mogącej być przyczyną bólu.
- Przyczyny: dysfunkcje w zakresie twarzy i żuchwy, lęk, depresja, przedawkowanie leków p/bólowych.
- Leczenie: niesteroidowe leki p/zapalne, miorelaksacyjne, p/depresyjne
46. Migrena – patogeneza, obraz kliniczny, rozpoznanie, leczenie
Migrena:
- Częstość: 10-15 % w populacji generalnej
- Kobiety chorują 3 x częściej niż mężczyźni populacji K> M (3:2, 4:1)
- Początek: przed 25 r. życia ( 25% w dzieciństwie)
- Etiopatogeneza:
- Dziedziczenie autosomalnie dominujące
- teoria naczyniowa
- teoria neurogenna
- teoria biochemiczna
Migrena bez aury:
- Wystąpienie co najmniej 5 napadów
- Częstość: pojedyncze do 2 tygodniowo
- Czas trwania napadu 4-72 godz.
- Napad ma przynajmniej 2 z wymienionych cech: ból po jednej stronie, pulsujący charakter bólu, średnie lub znaczne nasilenie bólu, wysiłek fizyczny nasila ból
- W czasie napadu występują nudności lub wymioty, fotofobia, fonofobia, osmofobia
Spełniony jest jeden z warunków:
- Brak innej choroby powodującej bóle
- Istnieje choroba ale nie jest powiązana czasowo z migreną
Migrena z aurą:
- 10-15% przypadków
- AURA :Zaburzenia korowe (mroczki, niedowidzenie połowicze, parestezje, drętwienia), niedowład połowiczy lub porażenie, afazja
- Faza bólowa
Migrena podstawna:
Spełnia warunki migreny z aurą
Występuje przynajmniej 2 z wymienionych objawów:
- niedowidzenie połowicze jednoimienne
- zaburzenia mowy typu dyzartrii
- zawroty głowy
- szum w uszach lub upośledzenie słuchu
- podwójne widzenie
- zaburzenia koordynacji ruchowej
- parestezje kończyn (mogą być obustronne)
- niedowłady kończyn (mogą być obustronne)
- zaburzenia świadomości
Zasady leczenia:
- Próba zidentyfikowania czynnika wywołującego ból głowy i jego eliminacja.
- Edukacja pacjenta
- Doraźne zwalczanie bólu
- Napady lekkie: nienarkotyczne środki p-bólowe (paracetamol, kwas acetylosalicylowy (1000 mg), metamizol, diklofenak, naproksen, leki przeciwwymiotne, uspokajające
- Napady średnio ciężkie i ciężkie: tryptany (50-100mg doustnie, 6mg i.m., 10-20 mg donosowo), ergotamina (0,25mg)
- Leczenie profilaktyczne (blokowanie receptorów 5-HT2): pochodne ergotaminy, leki przeciwserotoninowe, przeciwpadaczkowe, przeciwdepresyjne, beta-blokery.
- Akupunktura, fizjoterapia, uregulowany tryb życia.
47. Neuralgia nerwu V, obraz kliniczny, leczenie
Neuralgia nerwu trójdzielnego (Tic Douloureux) – jest nerwobólem jednej lub więcej gałęzi nerwu trójdzielnego, który jest V nerwem czaszkowym. Klasyczna neuralgia trójdzielna jest jednostronna i charakteryzuje się krótkotrwałymi, nagłymi napadami bólu w obrębie zakresu unerwienia jednej lub więcej gałązek nerwu, występującymi samoistnie lub po podrażnieniu tzw. stref spustowych.
Obraz kliniczny
Wiodącym objawem są napady bólu w obrębie połowy twarzy, w zakresie unerwienia nerwu V. Początek bólu jest nagły, a czas trwania wynosi od kilku sekund do maksymalnie 2 minut. Choroba ma charakter przewlekły, przebiega z okresami zaostrzeń i remisji.
Różnicowanie
W diagnostyce różnicowej neuralgii trójdzielnej powinno się uwzględnić:
- stwardnienie rozsiane
- tętniak tętnicy podstawnej mózgu
- guzy kąta mostowo-móżdżkowego
- konflikt naczyniowo-nerwowy
- zawał pnia mózgu
- jamistość opuszki.
Leczenie
Możliwości lecznicze neuralgii obejmują:
- farmakoterapię: lekiem z wyboru jest karbamazepina, inne stosowane leki to okskarbazepina, fenytoina, kwas walproinowy, topiramat, lamotrygina, gabapentyna, baklofen, klonazepam
- leczenie operacyjne: blokady gałązek nerwu, mikrochirurgiczne odbarczenie korzenia nerwu w przypadku konfliktu naczyniowo-nerwowego; stereotaktyczne zniszczenie przy pomocy promieni gamma.
48. Objawowe bóle głowy, przyczyny, różnicowanie
- Guzy mózgu
- Choroby naczyniowe oun np. krwotok podpajęczynówkowy, krwotok mózgowy
- Nadciśnienie tętnicze
- Choroby zakaźne gorączkowe
- Obejmujące ukł. nerwowy (zapalenie opon i mózgu)
- Ogólnoustrojowe (np. grypa)
- Choroby narządów twarzoczaszki
- Choroby krwi (czerwienica, białaczka)
- Choroby endokrynologiczne (nadczynność gruczołu tarczowego)
- Choroby kości czaszki
- Odległe skutki urazów
- Depresja i zespoły depresyjne
Mechanizmy bólów głowy:
- Ból głowy z pociągania - na skutek przemieszczenia i pociągania : zatok żylnych, dużych żył, opon np. guzy, krwiaki
- Ból głowy naczyniowy- nadmierne rozszerzenie bogato unerwionych naczyń : migrena, naczynioruchowy ból głowy, zatrucia.
- Nerwobóle w zakresie głowy i twarzy- nerwoból nerwu trójdzielnego
- Ból głowy mięśniowy-wskutek przykurczu odruchowego lub nadmiernego napięcia mięśni: napięciowy ból głowy, wady refrakcji, zaburzenia mięśni żucia
- Ból głowy rzutowany: jaskra, choroby uszu, zębów
49. Postępowanie diagnostyczne w bólach głowy
- Wywiad: wiek występowania, charakter bólu, częstość występowania, nasilenie bólu, umiejscowienie, występowanie rodzinne, wywiad rodzinny, choroby przebyte i aktualne, używki, leki
- Badanie stanu ogólnego i neurologiczne
- Odchylenia w stanie neurologicznym (CT, MRI, EEG)
- Stan neurologiczny prawidłowy: nie ma potrzeby badań za wyjątkiem sytuacji gdy:
- Istnieje podejrzenie padaczki lub guza
- Ból głowy jest odmienny niż poprzednio
- Ból jest nietypowy, bez reakcji na leki
Objawy alarmowe:
- Nagły, pierwszy w życiu ból głowy
- Najsilniejszy ból głowy jaki był w życiu
- Ból głowy po wysiłku
- Nowy ból głowy w wieku podeszłym
- Ból głowy narastający w ciągu kilku dni
- Obecność gorączki i/lub sztywności karku
- Obecność innych objawów neurologicznych (drgawki, niedowład, , zaburzenia mowy )
- Tarcza zastoinowa
- Ból głowy połączony z zaburzeniami świadomości
- Ból głowy u osoby z rakiem narządowym
50. Zawroty głowy, podział, przyczyny
Zawroty głowy: Subiektywne uczucie pozornego ruchu otoczenia lub własnego ciała, z niemożnością utrzymania odpowiedniej pozycji ciała.
Zespół ośrodkowy
- zawrót o typie zataczania, zapadania się, niepewności statycznej
- objawy narastają
- ruchy głowy nie mają wpływu na zawrót głowy
- nasilenie jest na stałym poziomie lub zmienne
- wymioty są rzadko
- objawy trwają miesiące lata
- rzadko występują zaburzenia słuchu
- występuje: podwójnie widzenie, przemijająca ślepota, zaburzenia świadomości, czasami drgawki
- objawy uszkodzenia oun
- przy próbie Romberga chory pada w kierunku ogniska.
Zespół obwodowy
- zawrót zawsze o typie wirowania
- nagły początek
- ruchy głowy nasilają zawrót głowy
- są największe na początku
- stopniowo wygasają
- występują wymioty
- trwają kilka, kilkanaście dni
- współistnieje szum w uchu
- upośledzenie słuchu
- nie ma zaburzeń wzrokowych, zaburzeń świadomości, drgawek, objawów ogniskowych
- przy próbie Romberga jest padanie w kierunku chorej przedsionka.
51. Układowe zawroty głowy, przyczyny, obraz kliniczny, leczenie
52. Nieukładowe zawroty głowy, przyczyny, obraz kliniczny, leczenie
53. Postępowanie diagnostyczne w zawrotach głowy
54. Co to jest padaczka
Padaczka jest to schorzenie przewlekłe, cechujące się skłonnością do występowania nawracających, nie prowokowanych napadów. O padaczce można mówić tylko wtedy, gdy u chorego wystąpiły dwa lub więcej samoistne napady padaczkowe w odstępie dłuższym niż 24 h . Pojedynczy napad padaczkowy uważany jest za zjawisko izolowane, nie spełnia więc kryteriów padaczki.
Napad padaczkowy jest wyrazem gwałtownego i nadmiernego wyładowania komórek nerwowych w wyniku depolaryzacji błony komórkowej. Towarzyszą temu zaburzeniu wyładowania czynności bioelektrycznej neuronów oraz objawy kliniczne zaburzeń ruchowych, zmysłowych, świadomości, emocjonalnych, wegetatywnych, poznawczych lub pamięciowych; często są to jednocześnie kombinacje tych zaburzeń.
55. Klasyfikacja napadów padaczkowych
Międzynarodowa klasyfikacja napadów padaczkowych (ICES)
- Napady częściowe
- Proste (z objawami ruchowymi, czuciowo-ruchowymi, autonomicznymi, psychicznymi)
- Złożone
- Napady częściowe wtórnie uogólniające się
- Napady uogólnione
Napady nieświadomości (typowe i atypowe)
Inne (miokloniczne, kloniczne, toniczne, toniczno-kloniczne, atoniczne)
56. Pierwotnie uogólnione napady padaczkowe
57. Napady padaczkowe częściowe
O napadzie częściowym mówimy, gdy objawy kliniczne wskazują, że napad rozpoczyna się w określonym obszarze kory mózgowej. Objawy podmiotowe i przedmiotowe występujące podczas napadów częściowych zależne są od okolicy mózgu, w której napad rozpoczyna się.
58. Etiopatogeneza padaczki
Padaczki zlokalizowane i uogólnione można jeszcze podzielić w zależności od prawdopodobnej etiologii napadów na idiopatyczne, objawowe i skrytopochodne
U 50% chorych nie udaje się znaleźć ewidentnej przyczyny
Mechanizm wyzwalający powoduje uszkodzenie struktury błony komórkowej neuronu z zaburzeniem jej funkcji i obniżeniem progu drgawkowego.
59. Przyczyny padaczki objawowej
Padaczka objawowa może być spowodowana chorobami naczyniowymi, zwłaszcza udarem, alkoholizmem, guzami mózgu, urazami głowy, zabiegami chirurgicznymi w obrębie półkul mózgu, przyczynami metabolicznymi (hiponatremia, hipernatremia, hipoglikemia, mocznica, uszkodzenie wątroby, ostra hipoksja, porfiria), chorobami zwyrodnieniowymi, deprywacją snu, wrażliwością na światło, przyczynami toksycznymi (amfetamina, leki przeciwdepresyjne, inhibitory MAO), odstawieniem leku.
Do uszkodzenia mózgu i padaczki u dziecka może dojść wskutek infekcji wewnątrzmacicznych, używania narkotyków przez matkę, naświetlania RTG we wczesnej ciąży, niedotlenieniem okołoporodowym
60. Leczenie padaczki
Przed rozpoczęciem leczenia farmakologicznego należy mieć pewność co do rozpoznania padaczki.
Zasady farmakoterapii: Długi czas leczenia. Leki przeciwpadaczkowe należy stosować przez długi okres, niekiedy do końca życia, schemat leczenia powinien więc być jak najprostszy.
Leczenie należy rozpocząć od stosowania jednego leku; u większosci chorych pozwoli to uzyskać kontrolę napadów i uniknąć działań niepożądanych. Konieczność zmiany leku lub leczenia skojarzonego zachodzi mniej niż u 30% pacjentów. Najniższa skuteczna dawka.
Uporczywe lub nawracające napady mogą być spowodowane niesystematycznym przyjmowaniem leku, złym wchłanianiem, niewłaściwym dawkowaniem lub zastosowaniem leku nieodpowiedniego dla danego chorego.
- karbamazepina,
- kwas walproinowy
- fenytoina
- etosuksymid
- okskarbazepina
- gabapentyna
- tiagabina
- topiramat
- wigabatryna
- lamotrygina
- lewetiracetam
Chirurgiczne leczenie padaczki:
Nie jest już uważane w terapii padaczki za ostateczność.
W wybranych przypadkach padaczki zlokalizowanej, a zwłaszcza u dzieci i młodzieży, zabieg operacyjny może przynieść znaczące korzyści, a nierzadko całkowite uwolnienie od napadów.
61. Stan padaczkowy, postępowanie w stanie padaczkowym
Lekiem pierwszego wyboru są benzodwuazepiny. Leczenie rozpoczynamy do 10 mg Diazepamu lub 2 mg klonazepamu podanego dożylnie w ciągu 3-4 min. Jeżeli dawkę musimy powtórzyć, następne wstrzyknięcie może być po ½ h; dawka klonazepamu może być zwiększona do 5-8 mg.
W pierwszej fazie (przepowiadającej) podajemy lorazepam, diazepam, midazolam, paraldehyd
W drugiej fazie, wczesnej, podać krótko działającą benzodiazepinę i.v.
W trzeciej fazie, w stanie ustabilizowanym, stosuje się duże dawki fenobarbitalu lub fenytoiny i.v., fosfenytoina i.m. (niezarejestrowana w Polsce), alternatywnie walproinian
W fazie oporności na leczenie (po 1 h) znieczulenie pacjenta i zastosowanie oddechu zastępczego, podaje się i.v. tiopental lub propofol, monitoruje się eeg
Jeśli nie podejmie się leczenia, jest stanem zagrażającym życiu. Wymaga resuscytacji, zapewnieniu drożności dróg oddechowych, dostarczenia tlenu do tkanek.
62. Padaczka a ciąża
- Około 0,3–0,4% wszystkich urodzeń pochodzi od matek chorych na padaczką, które w ponad 90% są urodzeniami zdrowych dzieci po prawidłowo przebiegającej ciąży i porodzie
- O ile u potomstwa kobiet chorych na padaczkę nie stwierdza się wad rozwojowych w 94–96% ciąż, to w populacji ogólnej ryzyko malformacji nie dotyczy 98% przypadków
- U większości kobiet nie stwierdza się wpływu ciąży na stan kliniczny padaczki i zmiana leczenia nie jest konieczna
- Nie ma pewności, czy większą częstotliwość krwawień z dróg rodnych u ciężarnych z padaczką, należy łączyć ze stosowaniem LPP. Natomiast potwierdzono, że noworodki matek chorujących na padaczkę częściej wykazują cechy opóźnienia rozwoju fizycznego, zwłaszcza w odniesieniu do wagi urodzeniowej poniżej 2500g. mniejszego obwodu głowy i niższej punktacji w skali Apgar
- Ważnym wskaźnikiem jest zmniejszenie stężeń i okresu półtrwania LPP w krwiobiegu matki wskutek zmian farmakokinetyki, które zachodzą pod wpływem zmienionego metabolizmu w okresie ciąży
- Całkowite ryzyko wad wrodzonych wskutek wewnątrzmacicznej ekspozycji na LPP wynosi około 6% i jest dwukrotnie wyższe u matek nie przyjmujących leków przeciw-padaczkowych, a chorujących na padaczkę.
- Politerapia prowadzona trzema lub więcej lekami zwiększa 2 – 4 krotnie prawdopodobieństwo wystąpienia wady, zwłaszcza wtedy, gdy utrzymuje się wysokie stężenie leku w surowicy krwi
- Mechanizmy teratogenności leków przeciwpadaczkowych nie są w pełni poznane
- Cechą wspólną dużych polekowych wad płodu, czyli deficytów fizycznych mających początek w okresie organogenezy, jest zaburzenie stanu zdrowia w stopniu, który wymaga interwencyjnego leczenia chirurgicznego
- Do najczęściej występujących wad dużych należą rozszczepy twarzoczaszki (wargi, podniebienia, szczęki),
- wady serca, wady rozwojowe cewy nerwowej (przepukliny oponowe, oponowo – mózgowo - rdzeniowe, mikrocefalia, anencefalia, wodogłowie), anomalie szkieletowe i anatomiczne niedrożności przewodu pokarmowego
- Wady małe, które mogą powstać również po zakończeniu organogenezy, występują przede wszystkim jako zmiany dysmorficzne twarzy (hiperteloryzm, zez, opadanie powieki, przerost wargi, nieprawidłowe osadzenie lub kształt nosa i uszu, krótka szyja)
- Opieka nad kobietą leczoną z powodu padaczki, planującą ciążę powinna rozpocząć się przed poczęciem
- Jest to niezwykle istotne gdy weźmiemy pod uwagę fakt, że wady układu nerwowego, serca powstają we wczesnym okresie embriogenezy od 28 do 42 dnia od pierwszego dnia ostatniego krwawienia miesięcznego.
63. Diagnostyka padaczki
Diagnostyka opiera się na obrazie klinicznym. Dodatkowo wykonuje się badania obrazowe głowy, eeg i badanie krwi. Różnicowanie z omdleniami, napadami rzekomopadaczkowymi (konwersja), przemijajacym atakiem niedokrwiennym, hipoglikemią.
64. Podział długotrwałych zaburzeń świadomości
Skala Glasgow:
Otwieranie oczu
- 4 punkty - spontaniczne
- 3 punkty - na polecenie
- 2 punkty - na bodźce bólowe
- 1 punkt - nie otwiera oczu
Kontakt słowny:
- 5 punktów - odpowiedź logiczna, pacjent zorientowany co do miejsca, czasu i własnej osoby
- 4 punkty - odpowiedź splątana, pacjent zdezorientowany
- 3 punkty - odpowiedź nieadekwatna, nie na temat lub krzyk
- 2 punkty - niezrozumiałe dźwięki, pojękiwanie
- 1 punkt - bez reakcji
Reakcja ruchowa:
- 6 punktów - spełnianie ruchowych poleceń słownych, migowych
- 5 punktów - ruchy celowe, pacjent lokalizuje bodziec bólowy
- 4 punkty - reakcja obronna na ból, wycofanie, próba usunięcia bodźca bólowego
- 3 punkty - patologiczna reakcja zgięciowa, odkorowanie (przywiedzenie ramion, zgięcie w stawach łokciowych i ręki, przeprost w stawach kończyn dolnych)
- 2 punkty - patologiczna reakcja wyprostna, odmóżdżenie (przywiedzenie i obrót ramion do wewnątrz, wyprost w stawach łokciowych, nawrócenie przedramion i zgięcie stawów ręki, przeprost w stawach kończyn dolnych, odwrócenie stopy)
- 1 punkt - bez reakcji
65. Ilościowe zaburzenia świadomości
Ilościowe zaburzenia świadomości: ograniczenie prawidłowej reakcji na docierające bodźce z zewnątrz:
- zaburzenia uwagi
- splątanie
- stupor
- zamroczenie - chwilowa utrata świadomości
- nieprzytomność - sopor (półśpiączka) i somnolencja (znaczna senność)
- śpiączka
Jakościowe - zachowana pełna świadomość, ale procesy świadomości ulegają zmianie:
- zespół majaczeniowy
- zespół urojeniowy
66. Przyczyny długotrwałych zaburzeń świadomości
Przyczyny mózgowe:
- padaczka
- uraz
- krwiak nadtwardówkowy
- krwiak podtwardówkowy
- krwotok podpajęczynówkowy
- krwiak śródmózgowy
- encefalopatia nadciśnieniowa
- zator w obszarze kręgowo-podstawnym
- zomr
- zapalenie mózgu
- ropień mózgu
Przyczyny pozamózgowe:
- kardiogenne: arytmie, zawał
- wstrząs septyczny
- hipowolemia
- hipo- lub hipernatremia
- hipo- lub hiperkalcemia
- hipo- lub hipertermia
- mocznica
- hipoksja
- niewydolność wątroby
- hiperkapnia
- przełom nadnerczowy
- niedoczynność przysadki
- obrzęk śluzowaty
- niedoczynność lub nadczynność przytarczyc
- toksyny
- alkohol
- CO
- leki
- przyczyny psychiatryczne: katatonia, śpiączka psychogenna
67. Badanie chorego nieprzytomnego
N - neck
A - drożność dróg oddechowych
B - oddychanie
C - krążenie
D - drugs, diabetes
E - epilepsy
F - fever
G - Glasgow
H - herniation
I - investigate
Badanie przedmiotowe:
- ewent. urazy głowy
- sztywność karku
- wzorzec oddechowy
- odruchy źreniczne
- ruchy gałek ocznych
- dno oka
- ułożenie kończyn, ruchy spontaniczne
- odruchy ścięgniste i podeszwowy
- punktacja w skali Glasgow
68. Badania dodatkowe w diagnostyce stanów nieprzytomności
- temperatura
- glikemia
- elektrolity
- morfologia krwi z rozmazem i ocena układu krzepnięcia
- gazometria krwi tętniczej
- posiew krwi
- toksykologia
- EKG
- RTG płuc
- obrazowanie mózgu
- płyn m-r
- EEG
- angiografia
69. Przyczyny omdleń
Termin omdlenie odnosi się do przejściowych zaburzeń przytomności , z towarzyszącą utratą napięcia mięśniowego, wynikających z nagłego, odwracalnego, globalnego spadku przepływu mózgowego.
Omdlenie z podrażnienia nerwu błędnego (wazowagalne)
- Pojawia się w ściśle określonych sytuacjach
- widok krwi (nawet w telewizji)
- nagły silny ból (uraz, kolka, leczenie zęba itd)
- długotrwałe przebywanie w pozycji stojącej
- przebywanie w gorącym, zatłoczonym pomieszczeniu
- nagłe przeżycie emocjonalne.
Niedociśnienie ortostatyczne
- Występuje w czasie wstawania po dłuższym przebywaniu w pozycji stojącej lub siedzącej, czasami po obfitym posiłku
Zaburzenia rytmu serca
- Rutynowe badanie ekg lub monitorowanie metodą Holtera pozwala potwierdzić rozpoznanie zaburzeń rytmu jako przyczyny omdleń.
Zjawisko Valsalvy
- Występuje w czasie codziennych zajęć, jak np. podczas podnoszenia ciężkich przedmiotów, defekacji, w czasie gry na instrumencie dętym itp. Może doprowadzić do omdlenia.
Choroby serca
- Przeszkoda w opróżnianiu lewej komory (zwężenie aorty, obturacyjna kardiomiopatia przerostowa)
- przeszkoda na poziomie zastawki przedsionkowo – komorowej
- ciężkie zaburzenia kurczliwości komór
Ostra niewydolność krążenia:
- niewydolność serca
- ostry zawał mięśnia sercowego
- zatorowość płucna
- tamponada serca
- niewydolność krążenia obwodowego (np. utrata krwi)
Omdlenie mikcyjne
Występuje w czasie, lub tuż po mikcji,
występuje w dwóch grupach wiekowych:
- u młodych mężczyzn, którzy wstają nagle w nocy chcąc oddać mocz
- u kobiet i mężczyzn w wieku podeszłym ze współistniejącymi schorzeniami układu krążenia i tendencją do ortostatycznych spadków ciśnienia.
Omdlenie wywołane defekacją:
- Zdarza się u osób w wieku podeszłym, cierpiących na zaparcia, ze współistniejącą chorobą niedokrwienną serca i ortostatycznymi spadkami ciśnienia.
Związane z nadwrażliwością zatoki szyjnej:
- Kilkusekundowy masaż zatoki szyjnej (poniżej kąta żuchwy) wywołuje zwolnienie czynności serca (z asystolią trwającą ponad 3 sek.), czasami również spadek skurczowego ciśnienia tętniczego (o ponad 50 mm Hg).
Przy połykaniu
- Przełykanie może w mechanizmie odruchu z nerwu błędnego wywołać bradykardię, blok przewodzenia przedsionkowo-komorowego i w konsekwencji omdlenie, rzadziej tachyarytmię.
W czasie jedzenia lodów
- Spożywanie zimnych pokarmów, zwłaszcza lodów, może w mechanizmie stymulacji nerwu błędnego prowadzić do omdlenia u dzieci, rzadziej u dorosłych
Kaszlowe
- Pojawia się w czasie uporczywych ataków kaszlu, np. u osób z przewlekłym nieżytem oskrzeli.
Łagodne omdlenie wieku młodzieńczego:
- Chłopcy w wieku młodzieńczym, wyjątkowo dziewczęta, mdleją niekiedy przy próbie wstania po przebywaniu przez godzinę lub dwie w pozycji siedzącej lub leżącej, zdarza się to zwłaszcza wieczorem. Omdlenia tego typu nie występują rano po obudzeniu.
70. Zaburzenia snu
- Bezsenność (insomnia)
- Nadmierna senność (hipersomnia)
- Zaburzenia rytmu snu (dyssomnia)
- Dysfunkcje przysenne (parasomnia)
- Niedobór snu (hiposomnia).
Formy bezsenności:
- utrudnienie zasypiania
- częste budzenie w ciągu nocy
- przedwczesne budzenie
- niewłaściwa jakość snu.
Nadmierna senność pierwotna
- Narkolepsja, zespoły snu z bezdechem, hipersomnia samoistna, zespół Kleinego-Levina, hipersomnia okołomiesiączkowa.
Nadmierna senność wtórna
- Niedobór snu, zaburzenia rytmu snu i czuwania, leki, alkoholizm, zatrucia zawodowe, choroby ogólnoustrojowe (niedokrwistość, niedoczynność tarczycy, niewydolność oddechowa), choroby organiczne układu nerwowego, depresja.
- Polega na występowaniu w ciągu dnia wielokrotnych napadów przymusowego, krótkotrwałego snu. Mogą dołączyć się inne objawy, jak katapleksja (napadowa utrata napięcia mięśniowego), przysenne porażenie mięśni oraz przysenne omamy.
71. Zjawiska przysenne (parasomnie)
Somnambulizm: Występuje u około 15% dzieci, na ogół mija w wieku dorosłym. Niekiedy ma związek z marzeniami sennymi. Dziecko, jeżeli zostanie nagle obudzone, wykazuje niewielkie zaburzenia orientacji.
Lęki nocne: Wyraźnie należy je oddzielić od zaburzeń z kręgu padaczki. Niektóre dzieci (wyjątkowo tylko dorośli) budzą się nagle w nocy z krzykiem, wyglądają na bardzo przerażone, nie poznają otoczenia, mogą mieć halucynacje. Rano istnieje całkowita niepamięć wydarzeń nocnych.
Bruksizm: Polega na zgrzytaniu zębami w czasie snu. Występuje u dzieci i bardzo rzadko u dorosłych.
Niekiedy wiąże się ze stresem lub urazem głowy.
Kiwanie głową: Kiwanie czy uderzanie głową w czasie snu występuje czasami u dzieci. Przyczyna nie jest znana.
Nocne mioklonie: Nocne „mioklonie” polegają na pojedynczych zrywaniach mięśniowych kończących się fazą toniczną , występujących w obrębie kończyn górnych i dolnych.
Porażenie przysenne: Polega na nagłej utracie możliwości poruszania się, połączonej z napadami lęku, często związanymi z treścią marzeń sennych. Napady te występują w chwili zasypiania, lub budzą chorego w nocy.
72. Napady pochodzenia mózgowego
73. Napady rzekomopadaczkowe psychogenne
Symptomatologia tych napadów może sugerować formy napadów częściowych prostych, złożonych i wtórnie lub pierwotnie uogólnionych. Napady mogą być drgawkowe i bezdrgawkowe, częściej występują u kobiet.
Podejrzewamy, gdy:
- Duża częstość napadów
- brak odpowiedzi na LPP
- emocjonalny rozrusznik
- obecność innych osób
- trudności zaobserwowania napadów przez personel
- wyjątkowo oddanie moczu
- brak przegryzienia języka
- brak właściwej reakcji chorego na częste napady
- znajomość padaczki
- częste pobyty w izbach przyjęć i hospitalizacje
- brak zmian w badaniach laboratoryjnych.
Objawy sugerujące zaburzenia konwersyjne
- Emocjonalny rozrusznik
- stopniowy początek
- nie „neurofizjologiczny”rozwój i przebieg napadu
- nieskoordynowana czynność ruchowa
- dystoniczne cechy
- wyrzucanie miednicy
- mięśnie twarzy nie włączone w napad uogólniony
- krzyk w czasie lub po napadzie
- stopniowe ustępowanie
- napad można zasugerować, prowokować lub przerwać.
74. Późne następstwa urazów
- pourazowy ropień mózgu
- padaczka pourazowa
- wodogłowie normotensyjne (zespół Hakima) - otępienie, ataksja, nietrzymanie moczu
- encefalopatia pourazowa
75. Objawy ogólne guzów śródczaszkowych
Objawy nadciśnienia śródczaszkowego
- Bóle głowy - wczesnoporanne, po przebudzeniu
- Nudności i wymioty zwykle w guzach podnamiotowych, na czczo
- Bradykardia
- Obrzęk tarcz nerwów wzrokowych
- Napady padaczkowe (uogólnione) najczęstszy objaw “ogólny” guza mózgu
- Objawy psychiczne apatia, senność, astenia funkcji psychicznych
Nagły wzrost ciśnienia śródczaszkowego
- Krwotok do guza
- Zawał w guzie
- Krwotok lub zawał w otoczeniu guza
- Krwotok do przestrzeni płynowych
- Ostre wodogłowie obturacyjne
Zespół wklinowania:
- Tożstronne rozszerzenie źrenicy
- Przeciwstronny niedowład połowiczy
- Zaburzenia przytomności
- Bradykardia
- Przeciwstronne rozszeżenie źrenicy
- Tożstronny niedowład połowiczy
- Sztywność odmóżdżeniowa
- Zaburzenia oddychania, potem krążenia
Wgłobienie mózgu to określenie stanu, w którym wzrost ciśnienia śródczaszkowego prowadzi do przemieszczenia części mózgowia z fizjologicznego przedziału anatomicznego do innego.
Śródczaszkowe przedziały anatomiczne wyznaczone są przez kości czaszki i wypustki opon: sierp mózgu oraz sierp móżdżku. Istnieją następujące możliwości wgłobień:
- wgłobienie zakrętu obręczy pod sierp mózgu, zazwyczaj nie ma uciśnięcia ważnych życiowo struktur (rzadko zamknięciu ulega tętnica okołospoidłowa); polega na wgłobieniu zakrętu obręczy do przestrzeni między dolnym brzegiem sierpu mózgu (zatoką strzałkową dolną) a górną powierzchnią ciała modzelowatego;
- wgłobienie we wcięcie namiotu (wklinowanie boczne, wklinowanie haka), gdy przyśrodkowa część płata skroniowego nazywana hakiem zakrętu przyhipokampowego przemieszcza się między wcięcie brzegu namiotu a śródmózgowie; następuje uciśnięcie tożstronego konaru mózgu, leżących we wcięciu namiotu nerwu okoruchowego i tętnicy tylnej mózgu oraz śródmózgowia. W niektórych przypadkach wgłobienia we wcięcie namiotu na konarze mózgu powstaje odwracalne wcięcie, określane jako wcięcie Kernohana (Kernohan's notch).
- wgłobienie w otwór wielki polega na przemieszczeniu migdałków móżdżku do otworu wielkiego. Może do niego dojść wskutek znacznego wzrostu ciśnienia śródczaszkowego, towarzyszącego niektórym guzom nadnamiotowym, i, przede wszystkim, w guzach tylnego dołu czaszki. Przemieszczenie pnia mózgu może doprowadzić do wtórnych wylewów krwawych do pnia, tzw. krwotoki Dureta.
- wgłobienie tylne (wgłobienie pokrywowe) - rzadko spotykana patologia, polegająca na ucisku tylnych części przyśrodkowych płatów skroniowych na wzgórki górne blaszki pokrywy
- wgłobienie środkowe (wgłobienie osiowe) polega na przemieszczeniu w otwór wielki całego pnia mózgu. Dochodzi do napięcia i rozerwania gałęzi przeszywających odchodzących od tętnicy podstawnej, zawału i krwotoku do pnia.
Wgłobienie zakrętu obręczy pod sierp mózgu:
Zazwyczaj jest nieme klinicznie. Udar niedokrwienny kory przyśrodkowej powierzchni półkuli mózgu i niedowład kończyny dolnej w wyniku zamknięcia tętnicy okołospoidłowej jest rzadkim powikłaniem. Rozpoznawane w badaniach neuroobrazowych jako przemieszczenie linii środkowej (midline shift).
Wgłobienie haka zakrętu przyhipokampowego we wcięcie namiotu móżdżku
- Uciśnięcie nerwu III spowoduje w pierwszej kolejności rozszerzenie źrenicy i brak reakcji na światło po stronie zmiany wywierającej ucisk. Uciśnięcie śródmózgowia powoduje kolejno:
- objawy uszkodzenia dróg piramidowych: niedowład połowiczy i objaw Babińskiego po stronie uszkodzenia w przypadku uciśnięcia konaru mózgu przez wcięcie namiotu; fałszywie lokalizujący objaw, określany też jako zespół Kernohana-Woltmana;
- zaburzenia przytomności;
- sztywność odmóżdżeniową z prężeniami;
- niewydolność krążeniowo-oddechową i śmierć.
Powikłaniem wklinowania we wcięcie namiotu może być ślepota korowa wywołana zawałem płatów potylicznych, powstałym w wyniku zamknięcia tętnic tylnych mózgu.
Wgłobienie migdałków móżdżku do otworu wielkiego
W przewlekłym wklinowaniu obserwowane są:
- sztywność karku
- przymusowe pochylenie głowy
- zaburzenia z nerwów czaszkowych
- objawy z długich szlaków nerwowych
- bradykardia
- czkawka
- niekiedy parestezje na karku i ramionach.
Ostre wklinowanie prowadzi do nagłej śmierci w związku z uszkodzeniem ośrodka oddechowego. Typowo występuje bezdech przy zachowanej świadomości pacjenta. Bradypnoë z częstością oddechów mniejszą niż 10/minutę często towarzyszy zagrażającemu wgłobieniu w otwór wielki.
76. Guzy siodła tureckiego i okołosiodłowe
- Dysfunkcje wewnątrzwydzielnicze
- Bóle głowy
- Zaburzenia pola widzenia (objawy późne)
- niedowidzenie dwuskroniowe
- ślepota jednooczna
- niedowidzenie skroniowe
- mroczki
- zanik nerwu lub nerwów wzrokowych
77. Guzy układu komorowego – napady Brunsa
Napady Brunsa:
Związane z nagłą zmianą pozycji głowy:
- Napadowe, silne bóle głowy
- Nudności i wymioty
- Bradykardia
- Zamącenie
78. Guzy kata mostowo-móżdżkowego
Zespół kąta mostowo-móżdżkowego
- Zanik słuchu, szumy, dzwonienie, dudnienie (faza słuchowa)
- Zawroty głowy, nudności, wymioty, zaburzenia równowagi (faza przedsionkowa)
- Uszkodzenie nerwu VII, V
Objawy późne
- tożstronna ataksja
- przeciwstronny niedowład połowiczy
- obustronny niedowład połowiczy
Zespół kąta mostowo-móżdżkowego to zespół objawów spowodowany obecnością guza w okolicy kąta mostowo-móżdżkowego. Najczęstszym guzem w tej okolicy są nerwiaki osłonkowe nerwu przedsionkowego, inne częste zmiany to oponiaki albo torbiel epidermoidalne, rzadkimi przyczynami są guzy przerzutowe, tętniaki, torbiele pajęczynówki, nerwiaki osłonkowe nerwu V.
Na obraz kliniczny zespołu składają się:
- objawy uszkodzenia nerwu VIII: zaburzenia słuchu, nieprzyjemne szumy uszne, zawroty głowy
- objawy uszkodzenia nerwu V: osłabienie lub brak odruchu rogówkowego, niedoczulica połowy twarzy
- objawy uszkodzenia nerwu VII: obwodowy niedowład (porażenie Bella)
- objawy ucisku na móżdżek i pień mózgu: tożstronne objawy móżdżkowe, przeciwstronne objawy piramidowe
- w zaawansowanym procesie występują również objawy uszkodzenia nerwów IX, X i XII i ciasnoty śródczaszkowej.
79. Guzy śródczaszkowe – algorytm diagnostyczny
- TK z kontrastem (EEG jeśli napady)
- MRI z kontrastem
- Angiografia RM (MRA) Angiografia DSA
- Biopsja stereotaktyczna (pierwotne chłoniaki mózgu) Badanie płynu m.-rdz.: poziom białka, cytologia (immunocytologia)
80. Guzy przedniego dołu czaszki i skrzydła mniejszego kości klinowej
Zespół rynienki węchowej/ skrzydła mniejszego
- Anosmia, zwykle bezobjawowa
Zespół płata czołowego
- nieadekwatne, głupkowate żarty (moria)
- abulia
- zamącenie
- Tożstronna ślepota i zanik nerwu II, ucisk przez masy guza
- Przeciwstronny obrzęk tarczy n. II, cechy ciasnoty śródczaszkowej
- Niekiedy elementy zespołu czołowego
81. Guzy kanału kręgowego – klasyfikacja kliniczna
Zewnątrzrdzeniowe
- nadtwardówkowe - raki, chłoniaki, szpiczaki mnogie
- podtwardówkowe - oponiaki, nerwiaki (55-60 %)
Śródrdzeniowe
- wyściółczaki (ependymoma myxopapillare)
- gwiaździaki
- skąpodrzewiaki
82. Guzy zewnątrzrdzeniowe – symptomatologia kliniczna
Bóle korzeniowe
- cięcie nożem (segmentowy)
- parestezje
- zaniki mięśniowe
- osłabienie (zanik) odruchów głębokich
Faza rdzeniowa
- częściowe uszkodzenie rdzenia (zespół Brown-Sequarda)
- całkowite (poprzeczne) uszkodzenie rdzenia
83. Guzy śródrdzeniowe – symptomatologia kliniczna
- Bóle
- Zespół syryngomieliczny
- rozszczepienne zaburzenia czucia
- zespół amiotroficzny (kończyny górne)
- niedowład spastyczny paraparesis (kończyny dolne)
- zaburzenia zwieraczy
- Częściowe lub całkowite uszkodzenie rdzenia
84. Guzy kanału kręgowego – algorytm diagnostyczny
- MRI z kontrastem
- potem TK/mielografia albo badanie pmr (próba Queckenstedta, białko, cytologia płynu)
- na końcu angiografia, DSA, MRA
Jeśli podejrzewamy, że uciśnięty jest rdzeń kręgowy, postępowanie diagnostyczne musi być przeprowadzone jak najszybciej, bowiem dekompensacja funkcji rdzenia może nastąpić nagle, w nie dającym się przewidzieć czasie.
85. Choroba Parkinsona, etiopatogeneza, obraz kliniczny
W chorobie Parkinsona do objawów chorobowych dochodzi z powodu zmian zwyrodnieniowych komórek nerwowych w istocie czarnej i innych obszarach barwnikonośnych mózgowia. Neurony istoty czarnej wytwarzają neurotransmiter dopaminę, stąd nazywa się je neuronami dopaminergicznymi; zawierają ponadto melaninę, stad określa się je jako barwnikonośne. Konsekwencją zaburzenia funkcji tych neuronów jest niedobór dopaminy (ok. 70-80%) w istocie czarnej i prążkowiu, i przewaga aktywności neuronów glutaminergicznych, hamujących jądra wzgórza. W zmienionych chorobowo obszarach mózgowia stwierdza się obecność ciał Lewy'ego, jednak nie są to zmiany patognomoniczne dla choroby Parkinsona.
Objawy neuropatologiczne korelują z obrazem klinicznym choroby.
W proces neurodegeneracyjny wciągnięte są układ dopaminergiczny (istota czarna, podwzgórze, układ mezokortykalno-limbiczny, siatkówka), układ noradrenergiczny (miejsce sinawe), układ cholinergiczny (jądro podstawne Meynerta) i układ glutaminergiczny.
Czynniki wywołujące te zmiany zwyrodnieniowe do chwili obecnej nie zostały dostatecznie określone, jednak przynajmniej część z nich to czynniki genetyczne.
Objawy:
Objawy prodromalne:
- sztywność osobowości
- depresja
- zaparcia
- zapalenie łojotokowe skóry
- parestezje kończyn
- dyskretne zaburzenia węchowe
Podstawowe objawy:
- drżenie (spoczynkowe)
- spowolnienie ruchowe
- wzmożone napięcie mięśniowe (sztywność)
- zaburzenia postawy
- zaburzenia chodu
- hypomimia
- utrata współruchów (np.balansowania kończyn)
- dyzartria
- mikrografia
Kryteria rozpoznania PD:
Rozpoznanie możliwe
- Postępujący przebieg i
- Przynajmniej 2 z 3 następujących objawów:
- akineza
- sztywność mięśni
- drżenie spoczynkowe
- brak cech nietypowych dla PD.
Rozpoznanie prawdopodobne
- Spełnione kryteria dla możliwej PD i
- Przynajmniej 2 z następujących objawów:
- wyraźna poprawa po lewodopie
- występowanie fluktuacji lub dyskinez w związku z leczeniem lewodopą
- asymetria objawów.
Rozpoznanie pewne
- Spełnione kryteria dla prawdopodobnej PD i
- Wykazanie w badaniu sekcyjnym:
- zaniku neuronów istoty czarnej
- obecności ciał Lewy'ego w istocie czarnej
- braku ciał wtrętowych w komórkach oligodendrogleju
86. Najważniejsze przyczyny zespołu parkinsonowskiego
Zespoły parkinsonowskie mogą być spowodowane czynnikami:
- toksycznymi - np. zatrucie talem, litem, związkami ołowiu, tlenkiem węgla,
- farmakologicznymi - ze względu na udział obwodów dopaminergicznych w powstawaniu parkinsonizmu oraz psychoz, możliwe jest wystąpienie parkinsonizmu polekowego w przewlekłych zaburzeniach psychicznych,
- naczyniowymi - miażdżyca, udar mózgu,
- ekspansywnymi śródczaszkowmi,
- urazowymi.
Parkinsonizm plus (parkinsonizm atypowy) – grupa pierwotnych schorzeń neurodegeneracyjnych charakteryzujących się obecnością objawów parkinsonowskich współistniejących z innymi objawami uszkodzenia układu nerwowego.
Najczęstszymi schorzeniami w grupie parkinsonizmu plus są:
- postępujące porażenie nadjądrowe (PSP)
- zanik wieloukładowy (MSA)
- zwyrodnienie korowo-podstawne (CBD)
- otępienie z ciałami Lewy'ego (LBD).
W diagnostyce parkinsonizmu-plus należy zwrócić uwagę na tzw. objawy ostrzegawcze, które sugerują rozpoznanie jednego ze schorzeń tej grupy a nie choroby Parkinsona. Są to:
- ograniczona odpowiedź na lewodopę
- objawy postawne (upadki)
- otępienie we wczesnym etapie choroby
- dysfagia, dyzartria, stridor, dyzautonomia we wczesnym okresie choroby
- objawy piramidowe
- objawy móżdżkowe
- uszkodzenie obwodowego neuronu ruchowego
- objawy zespołu płata czołowego
- objawy zespołu płata ciemieniowego
- palilalia, echolalia
- dystonia
- mioklonie
- zaburzenia ruchomości gałek ocznych w pionie
- wczesny, nagły początek choroby
- gwałtowna progresja
- symetryczny początek
- brak drżenia, drżenie nietypowe.
87. Leczenie wczesnej i zaawansowanej postaci choroby Parkinsona
- L-DOPA jest zasadniczym lekiem stosowanym w farmakoterapii PD.
- Amantadyna to lek zwiększający uwalnianie endogennej dopaminy.
- Agoniści receptorów dopaminowych, np.bromokryptyna.
- Inhibitory MAO hamujące rozkład dopaminy, np. selegilina.
- Inhibitory COMT (katecholo-o-metylotransferazy): entakapon, tolkapon, nitekapon.
- Leki antycholinoergiczne: biperiden, cykrymina, procyklidyna, triheksyfenidyl.
- Beta-blokery, np. Propranolol.
Niefarmakologiczne:
- muzykoterapia – przede wszystkim taniec
- leczenie operacyjne – stereotaktyczne uszkadzanie gałki bladej (pallidotomia), jądra niskowzgórzowego (subtalamotomia) albo jąder wzgórza (talamotomia), jedno- lub obustronne
- przeszczep płodowej istoty czarnej
- Głęboka stymulacja mózgu: wszczepienie elektrostymulatora w zidentyfikowane ognisko w mózgu.
88. Pląsawica Huntingtona – etiopatogeneza, obraz kliniczny, różnicowanie
Choroba ujawnia się w późnym wieku (na ogół u osób w wieku 35-50 lat). Młodzieńcza odmiana choroby (postać Westphala) dotyczy ludzi przed 20. rokiem życia (10% przypadków). Od momentu rozpoznania średni czas przeżycia wynosi 15-20 lat. Częstość występowania choroby Huntingtona szacuje się na 4-8:100 000
Przyczyną choroby jest mutacja w genie HD kodującym białko huntingtynę, położonym na chromosomie 4. Choroba dziedziczona jest w sposób autosomalny dominujący. Oznacza to, że statystycznie połowa potomstwa chorego na pląsawicę odziedziczy zmutowany allel powodujący chorobę. Zmutowany allel genu utrzymuje się w populacji ze względu na późne wystąpienie objawów choroby. Mutacja polega na ekspansji trójki nukleotydowej CAG (kodon oznaczający aminokwas glutaminę). Powoduje to, że w sekwencji aminokwasowej huntingtyny pojawia się długi ciąg glutamin. Jeśli powtórzeń trójki CAG jest więcej niż 35, mutacja staje się niestabilna, i przy kolejnych podziałach komórkowych ilość powtórzeń się zwiększa. W związku z tym w kolejnych pokoleniach ciąg glutamin w białku jest coraz dłuższy, a co za tym idzie, objawy choroby pojawiają się wcześniej i są silniejsze. Takie zjawisko nazywa się antycypacją. W chorobie Huntingtona ekspansja trójnukleotydów jest znacząco większa przy przekazie mutacji od ojca.
Nieprawidłowe białko gromadzi się w komórkach nerwowych, powodując ich śmierć. Przypuszcza się, że neurotoksyczność zmutowanej huntingtyny wiąże się z dysfunkcją mitochondriów, jednak dokładny mechanizm patogenezy nie jest znany. Zmiany dotyczą przede wszystkim jądra ogoniastego, skorupy i kory mózgowej.
Diagnozy dokonuje się na podstawie objawów klinicznych. W badaniach obrazowych (TK i MRI) stwierdza się poszerzenie układu komorowego mózgowia, z charakterystycznym obrazem komór bocznych, mających kształt "skrzydeł motyla".
Badania genetyczne mogą dać odpowiedź, czy pacjent ma zmutowany allel genu, także zanim wystąpią objawy choroby. Możliwe są również genetyczne badania prenatalne.
Do objawów HD należą:
- niekontrolowane ruchy (ruchy pląsawicze)
- pojawienie się drżenia rąk i nóg
- zmniejszenie napięcia mięśni (nie dotyczy postaci młodzieńczej, w której napięcie mięśni jest wzmożone)
- zaburzenia umysłowe, otępienie, postępujące zaburzenia pamięci
- zmiany osobowości, depresja.
Przebieg choroby jest powolny i postępujący. Z czasem pojawiają się zaburzenia mowy i połykania, a chory staje się uzależniony od pomocy innych ludzi. Zachłystowe zapalenie płuc spowodowane zaburzeniami połykania jest pierwszą przyczyną śmierci pacjentów z HD, odpowiadając za 85% zgonów
89. Drżenie samoistne, rozpoznanie, leczenie
- Kryteria podstawowe:
- Obustronne, pozycyjne lub kinetyczne (nie spoczynkowe), drżenie kończyn górnych
- Brak innych objawów neurologicznych
- Odosobnione lub współistniejące drżenie głowy, bez cech dystonii
- Kryteria dodatkowe:
- Czas trwania objawów > 3 lata
- Dodatni wywiad rodzinny
- Ustępowanie zaburzeń po spożyciu alkoholu.
Rozpoznanie jest potwierdzone przy spełnionych kryteriach podstawowych, kryteria dodatkowe mają znaczenie pomocnicze (występują u >50% chorych).
Kryteria rozpoznania:
- widoczne, utrzymujące się, obustronne lub kinetyczne drżenie dłoni i/lub przedramion izolowane drżenie głowy, przy braku zaburzeń postawy
Kryteria wyłączenia:
- obecne inne nieprawidłowości neurologiczne
- współistnienie schorzeń mogących powodować nasilone drżenie fizjologiczne
- ewidentnie nagły początek i postępujący przebieg drżenia
- pierwotne drżenie ortostatyczne
- izolowane drżenie głosu, języka i brody, nóg,
- izolowane drżenie specyficzne dla pozycji lub zadania
- współistnienie czynników psychogennych
90. Dystonie – podział, przyczyny, obraz kliniczny, postępowanie terapeutyczne
Dystonia to dynamiczne zaburzenia napięcia mięśniowego z mimowolnymi przetrwałymi skurczami mięśni. Może być oddzielną jednostką chorobową jak i objawem choroby, może być uogólniona lub ogniskowa
Dystonie genetycznie uwarunkowane:
- Idiopatyczna dystonia torsyjna DYT1
- Idiopatyczna dystonia torsyjna DYT2
- Dystonia związana z chromosomem X-DYT3
- Dystonia reagująca ne Levodopę (DYT5 i DYT6)
- Dystonia z głuchotą
Ogniskowa
- (kurcz powiek, kręcz karku, skurcz twarzy, połowiczy kurcz twarzy)
- dystonia gardłowa obejmująca mięśnie krtani i gardła
Segmentalna ( np. objemująca sąsiednie okolice ciała np. szyję i kończynę górną lub mięśnie okrężne oka, żuchwę, wargi, język tzw. zespół Meige`a) .
Uogólniona obejmująca kilka różnych okolic ciała
Uogólniona obejmująca całe ciało
91. Tiki – obraz kliniczny, rozpoznanie, leczenie
Tiki to nagle występujące zjawiska ruchowe lub dźwiękowe przebiegające stereotypowo, ale pozbawione cech rytmiczności.
- Proste tiki motoryczne: mruganie oczami, marszczenie czoła, wyciaganie języka, otwieranie ust, potrząsanie głową.
- Proste tiki wokalne: kaszel, pochrząkiwanie, krzyk, szczekanie
- Złożone tiki motoryczne: zgrzytanie zębami, „strzelanie” stawami palców, odruchy klaskania, echopraksje, kopropraksje
- Złożone tiki wokalne: wydawanie więcej niż jednego dźwieku, koprolalia, echo/palilalia
Przejściowy zespół tików
- występuje przed 18 r.ż
- tiki pojedyncze lub złożone wielokrotnie w ciągu doby, codziennie, nie krócej niż 4 tyg. i nie dłużej niż 12 miesięcy
- wykluczenie przyczyn objawowych
Przewlekłe tiki motoryczne lub wokalne
- objawy trwają ponad 1 rok
Zespół Gilles de la Touretta
Zespół Gilles de la Tourette'a:
- wystąpienie złożonych tików ruchowych i wokalnych, co najmniej jednorazowo, przerwy nie dłuższe niż 3 miesięcy
W zespole Tourette'a w patogenezę zaangażowane jest jądro ogoniaste
Leczenie: haloperidol, inne neuroleptyki
92. Rozpoznanie różnicowe drżenia
Drżenie to rytmiczn ruch oscylacyjny dowolnej części ciała. Dzieli się je na wolne (<6 Hz) i szybkie (>6 Hz).
Drżenie
- fizjologiczne
- patologiczne
- drobnofaliste
- grubofaliste
- drżenie spoczynkowe w chorobie Parkinsona
- drżenie fizjologiczne
- drżenie samoistne
- drżenie zamiarowe w uszkodzeniu móżdżku
- drżenie pochodzenia czerwiennego - grubofaliste, wyjątkowo nasilone, ataktyczne, częscto w SM, nie reaguje na leki
93. Kurczowy kręcz karku – przyczyny, obraz kliniczny, leczenie
Dystonia szyjna
Objawy:
- hyperkinezy dotyczące mięśni szyi i karku, powodujące skręt szyi do boku (laterocollis), pochylenie głowy do przodu (anterocollis) lub do tyłu (retrocollis) wraz z drżeniem, tzw, drżenie dystoniczne - przerywane ruchy zrywania
- rekcja zatrzymania - powstrzymywanie ruchów mimowolnych ręką
- proces obejmuje mięsień moskowo-obojczykowo-sutkowy, górną część m.czworobocznego i niekiedy mięśnie pochyłe
Występowanie: wiek średni (3.-5. dekada życia)
Leczenie: toksyna botulinowa, rizotomia C1-4, akupunktura, odp. na leki antycholinergiczne często nieodpowiednia
94. ALS, etiopatogeneza, obraz kliniczny
Stwardnienie zanikowe boczne to nieuleczalna, postępująca choroba neurodegeneracyjna, która prowadzi do niszczenia komórek rogów przednich rdzenia kręgowego, jąder nerwów czaszkowych rdzenia przedłużonego oraz neuronów drogi piramidowej, czyli wybiórczego uszkodzenia obwodowego (dolnego) i ośrodkowego (górnego) neuronu ruchowego.
Pojawia się głównie w 6. i 7. dekadzie życia, częściej u mężczyzn. Częstość występowania choroby szacuje się na 2-4:100 000. Choroba może wystąpić też u młodych ludzi, chociaż zdecydowanie rzadziej (1-3:500 000)
Istnieją dwa typy ALS:
- uwarunkowane genetycznie ALS (FALS). Przyjmuje się, że do 20% przypadków stwardnienia bocznego zanikowego jest spowodowane mutacją genu kodującego dysmutazę ponadtlenkową (SOD1) na chromosomie 21.
- pozostałe przypadki to sporadyczne ALS.
Oba przypadki klinicznie nie są możliwe do rozróżnienia, ale zazwyczaj forma uwarunkowana genetycznie pojawia się we wcześniejszym wieku.
Wykrycie zależności pomiędzy FALS a mutacją genu SOD1 pozwoliło wysnuć przypuszczenie, że etiologia ALS może być ściśle powiązana z niedoborem (nadmierną) aktywności(-ą) dysmutazy ponadtlenkowej.
Istnieją dwie istotne hipotezy, które mogą mieć znaczący wpływ na poznanie patofizjologii ALS. Pierwsza hipoteza zakłada zyskanie funkcji (ang. gain of function). Według tej hipotezy patogeneza choroby jest związana z uzyskaniem nowych zdolności katalitycznych dysmutazy ponadtlenkowej, która wskutek zmian genetycznych posiada inną budowę trzeciorzędową (dysmutaza ponadtlenkowa jest metaloproteiną).
Druga hipoteza zakłada zyskanie zdolności do nowych interakcji (ang. gain of interction). Ta hipoteza przedstawiła zmutowane białko SOD1 jako nieaktywne katalitycznie ulegające szybkiej degradacji.
Jest możliwe by obie hipotezy były prawdziwe i się uzupełniały. Mechanizm wolnorodnikowy oraz konformacyjny spowodowane obiema hipotezami odrębnie nie muszą się wykluczać. Obecne badania wskazują, że główną rolę w rozwoju ALS powoduje zmiana konformacyjna SOD1, a co za tym idzie, utrata kontroli oddziaływań międzybiałkowych i tworzenie nierozpuszczalnych agregatów w komórkach (zawierających amyloid-β - charakterystyczny składnik złogów podejrzewany o generowanie wolnych rodników).
Ciągła i postępująca degeneracja motoneuronów w ALS prowadzi ostatecznie do ich śmierci, a co za tym idzie do utraty przez mózg możliwości kontrolowania pracy mięśni.
Pierwszymi objawami są zaniki mięśniowe dotyczące mięśni krótkich rąk, spastyczny niedowład kończyn dolnych oraz niekiedy cechy zespołu opuszkowego. W dalszym przebiegu pojawiają się zaniki kolejnych grup mięśniowych z charakterystycznymi drżeniami pęczkowymi oraz progresją objawów spastycznych. U pacjentów w toku rozwoju choroby dochodzi do powolnego, ale systematycznego, pogarszania się sprawności ruchowej, a w późniejszych etapach do całkowitego paraliżu i ostatecznie śmierci poprzez zatrzymanie pracy mięśni oddechowych. Ogólnie przyjmuje się, że ALS prowadzi do śmierci w przeciągu 3-5 lat; najczęściej śmierć z powodu ALS następuje wskutek niewydolności oddechowej (paraliż mięśni oddechowych). Poza nielicznymi przypadkami ALS nie dotyka sfery intelektualnej człowieka, przykładem może być astrofizyk, prof. Stephen Hawking, który walczy z chorobą od ponad 40 lat.
Klinicznie wyróżnia się niekiedy dwie początkowe postaci choroby:
- postać z objawami zlokalizowanymi w kończynach ("postać kończynową", ang. limb onset, około 75%)
- postać z cechami zespołu opuszkowego: zaburzeniami mowy, połykania ("postać opuszkową", ang. bulbar onset, około 25%).
Rozpoznanie opiera się na kryteriach World Federation of Neurology (kryteriach Airlie House/ El Escorial Revisited)
Rozpoznanie ALS wymaga
- A — obecności:
- A1 — uszkodzenia dolnego neuronu ruchowego (w badaniu klinicznym, EMG lub neuropatologicznym)
- A2 — uszkodzenia górnego neuronu ruchowego (w badaniu klinicznym)
- A3 — postępujących zaburzeń ruchowych w danym obszarze lub pojawienia się ich w innych obszarach (na podstawie wywiadu lub w badaniu klinicznym)
oraz
- B — wykluczenia:
- B1 — innych jednostek chorobowych, tłumaczących uszkodzenie górnego lub dolnego neuronu ruchowego (w badaniu EMG lub neuropatologicznym)
- B2 — innych jednostek chorobowych, mogących tłumaczyć zaburzenia kliniczne i elektrofizjologiczne (w badaniu neuroobrazowym: TK lub MRI).
Rozpoznanie różnicowe
- objawowe ALS (zespół paranowotworowy)
- uszkodzenie na poziomie opuszki (guz, jamistość opuszki, uszkodzenia naczyniowe)
- uszkodzenie na poziomie szyjnego odcinka rdzenia kręgowego (mielopatia szyjna, guz rdzenia)
- stwardnienie rozsiane
- wieloogniskowa neuropatia ruchowa
- zapalenie mięśni z ciałami wtrętowymi
- zatrucia metalami ciężkimi
95. Definicja otępienia, podział zespołów otępiennych
Otępienie jest zespołem spowodowanym chorobą mózgu, zwykle przewlekłą i postępującą. Charakteryzuje się zaburzeniami wyższych czynności korowych: pamięci, myślenia, orientacji, rozumienia, liczenia, zdolności do uczenia się, porównywania, oceniania i dokonywania racjonalnych wyborów. Towarzyszy mu osłabienie kontroli nad reakcjami emocjonalnymi, zachowaniem społecznym i motywacją. (ICD-10)
Typy otępienia uwzględniający lokalizację zmian
Korowe
- Choroba Alzheimera
- Otępienie czołowo-skroniowe
Podkorowe
- Pląsawica Huntingtona
- Postępujące porażenie nadjądrowe
- Choroba Parkinsona
- Choroby zapalne (AIDS)
- Stwardnienie rozsiane
- Choroba z obecnością ciałek Lewy’ego
Korowo-podkorowe (globalne)
- Otępienie naczyniowe
- Urazy czaszkowo-mózgowe
- Guzy mózgu
- Encefalopatie gąbczaste (choroba Creutzfeldta-Jakoba)
- Endokrynopatie
- Awitaminozy
- Toksyny (choroba Wilsona, alkohol)
Częstość:
- Choroba Alzheimera 40 %
- Otępienie naczyniowe 35 %
- Otępienie mieszane 10-15 %
- Inne zespoły otępienne 10-15 %
96. Choroba Alzheimera, patogeneza, symptomatologia kliniczna, różnicowanie
Uszkodzenie układu cholinergicznego
Hipokamp, j. migdałowate, j. Meynerta, kora nowa
Przewaga układu GABA-ergicznego
Wykładniki morfologiczne choroby Alzheimera
• zwyrodnienie włókienkowe typu Alzheimera -
pasma włókien z hiperfosforylowanych białek cytoskeletonu (MAP-tau)
• dystroficzne neuryty (DN) - paired helical filaments
• płytki neurytyczne - beta-amyloid, Apo E- 4, DN, PrP
97. Otępienie wielozawałowe
- zaburzenia pamięci świeżej
- zaniedbywanie się
- zaburzenia czynności wzrokowo-przestrzennych i wzrokowo-ruchowych
- afazja, apraksja
- odwrócenie cyklu dobowego czuwania
- nagły początek, zmienny przebieg z zaostrzeniami
- ogniskowe objawy neurologiczne
- obciążenie chorobami naczyniowymi: zmiany w ekg, rtg kl. piersiowej, dnie oka, lipidogramie
98. Otępienie w następstwie udarów w strefach strategicznych
99. Otępienie zatokowate
Cechy otępienia zatokowatego:
- upośledzenie pamięci świeżej
- brak inicjatywy (zainteresowań)
- spowolnienie spontanicznej aktywności umysłowej
- labilność emocjonalna
- dość sprawne zachowania społeczne
- wywiad „naczyniowy”, skryty początek i wolny,postępujący przebieg
- porażenie rzekomoopuszkowe,
- zaburzenia chodu, nietrzymanie moczu
100. Podkorowa encefalopatia miażdżycowa Binswanera
Leukoencefalopatia podkorowa (choroba Binswangera) to rzadko rozpoznawana postać otępienia wielozawałowego. Na obraz kliniczny choroby Binswangera składają się zespół otępienny, objawy ogniskowe, częste są napady padaczkowe. Niemal wyłącznie dotyczy pacjentów z nadciśnieniem tętniczym; częstsza jest u mężczyzn. W tomografii komputerowej stwierdza się symetryczne ogniska hipodensyjne w istocie białej.
Cechy choroby Binswangera
- otępienie typu podkorowego (jak zatokowate)
- wywiad „naczyniowy”, nadciśnienie tętnicze z epizodami istotnego spadku ciśnienia
- skryty początek
- wolny, postępujący przebieg
- porażenie rzekomoopuszkowe, zaburzenia chodu,
- nietrzymanie moczu
- Patologia długich naczyń tętniczych: zwłóknienie ściany, martwica ściany
- demielinizacja, uszkodzenie aksonów, ubytek oligodendrogleju
- drobne ogniska martwicze, niekiedy w jądrach podstawy
- przerost astrogleju
- leukoarajoza - niedokrwienie „ostatniej łączki”
101. Choroba Creutzfeldta-Jakoba
- Otępienie szybko postępujące
- Fluktuacje objawów, do mutyzmu włącznie
- Zespół pozapiramidowy
- Objawy piramidowe
- Zespół móżdżkowy
- Zespół amiotroficzny (zanik neuronów rogów przednich rdzenia kręgowego)
- Charakterystyczny zapis eeg (rytmiczne trójfazowe zespoły fal ostrych)
- Białko 14-3-3 w płynie mózgowo-rdzeniowym
102. Diagnostyka i leczenie otępienia naczyniowego
- Otępienie niedokrwienne
- otępienie wielozawałowe (MID)
- udar w strefie strategicznej
- choroba Binswangera
- Otępienie „hypoperfuzyjne”
- Otępienie pokrwotoczne
103. Choroba Alzheimera – postępowanie terapeutyczne
Leczenie niefarmakologiczne
- informowanie o chorobie
- rytm dnia, ruch
- kontakty interpersonalne
- dialog z chorym (polecenia)
- praca indywidualna i terapia zajęciowa
- domy opieki
Leczenie farmakologiczne
- inhibitory cholinesterazy (donepezil - Donepex, rywastigmina - Exelon, galantamina - Reminyl)
- leki nootropowe, np. piracetam
- antagoniści NMDA: memantyna (Axura)
- leki przeciwdepresyjne i przeciwlękowe
104. Objawy kliniczne miastenii
Skargi pacjentów
- Opadanie powiek (ptoza) – jednostronne, obustronne, naprzemiennie
- Rozdwojenie obrazu
- Zmiana mimiki twarzy, tzw. uśmiech poprzeczny
- Opadanie żuchwy
- Osłabienie gryzienia, żucia, połykania pokarmów, nasilające się w miarę jedzenia
- Osłabienie głosu, niewyraźna mowa w miarę wydawania głosu (apokamnoza)
- Opadanie głowy
- Osłabienie mięśni rąk w czasie mycia zębów, czesania włosów, golenia się
- Osłabienie siły mięśni nóg przy chodzeniu, upadki podczas biegu
- Zaburzenia oddychania
- Długi okres niepodejmowania oddechu po narkozie z użyciem kurary
Miastenia - grupy kliniczne:
- oczna
- opuszkowa
- uogólniona
- uogólniona z zajęciem mięśni oddechowych
Zmodyfikowana klasyfikacja Ossermana
- Grupa I - miastenia oczna
- Grupa IIA - łagodna miastenia uogólniona
- Grupa IIB - umiarkowana do ciężkiej miastenia uogólniona
- Grupa III - miastenia ostra (gwałtowna) lub ciężka uogólniona z niewydolnością oddechową
- Grupa IV - miastenia, późna, ciężka, ze znaczącą symptomatologią opuszkową
105. Badania dodatkowe przydatne w diagnostyce miastenii
- próba z chlorkiem edrofonium (Tensilonem): zmniejszenie lub zniesienie objawów po dożylnym podaniu 10 mg leku
- badania elektrofizjologiczne: stwierdzenie tzw. dekrementu miastenicznego w elektrostymulacyjnej próbie nużliwości (repetitive nerve stimulation test, RNS), ocena wyniku badania elektromiograficznego pojedynczego włókna mięśniowego (SFEMG, single-fiber electromyography).
- badanie przeciwciał anty-AChR i (lub) anty-MusK
- tomografia komputerowa śródpiersia, MRI
106. Leczenie miastenii
Leczenie farmaceutyczne ma na celu zwiększenie stężenia acetylocholiny w złączach nerwowo-mięśniowych i zapobieganie wytwarzania przeciwciał receptorów acetylocholiny. W pierwszym celu stosuje się inhibitory cholinoesterazy:
- neostygminę
- pirydostygminę
- ambenonium.
Produkcję przeciwciał ogranicza się leczeniem immunosupresyjnym. Stosowane leki to:
- prednizon, metyloprednizon
- azatiopryna
- cyklofosfamid
- metotreksat
- mykofenolan mofetylu.
Niekiedy stosuje się plazmaferezę i dożylne wlewy immunoglobulin.
Leczenie chirurgiczne polega na usunięciu grasicy (tymektomia), do czego bezwzględnym wskazaniem jest podejrzenie grasiczaka.
107. Nabyte zaburzenia transmisji nerwowo-mięśniowej
Wrodzone zaburzenia transmisji nerwowo-mięśniowej:
- defekt syntezy lub mobilizacji acetylocholiny
- niedobór acetylocholiesterazy płytkowej
- niedobór receptorów acetylocholiny
- zespół powolnego kanału receptora acetylocholiny
Nabyte zaburzenia transmisji nerwowo-mięśniowej:
Toksyczne:
- polekowe
- botulizm
- zatrucie pestycydami
Immunologiczne:
- miastenia
- zespół Lamberta-Eatona
- zespół po leczeniu cuprenilem
108. Podział i objawy kliniczne miopatii
Miopatie to choroby mięśni. Charakteryzuje je osłabienie mięśni, zwykle dosiebnych, każda miopatia ma szczególny wzorzec zajętych mięśni co jest istotne w diagnostyce.
Miopatie dziedziczne:
- dystrofie mieśniowe
- choroby miotoniczne
- miopatie metaboliczne
- porażenia okresowe
- miopatie mitochondrialne
Miopatie nabyte:
- miopatie zapalne
- miopatie endokrynologiczne i metaboliczne
- miopatie polekowe
- miopatie paranowotworowe
- miopatie stanu krytycznego
109. Miopatie nabyte – przyczyny
- zapalne (infekcyjne, autoimmunologiczne)
- metaboliczne/ endokrynologiczne
- toksyczne
Miopatie zapalne autoimmunologiczne
- PM- polymyositis
- DM- dermatomyositis
- IBM- inclusion body myositis
Występują jako
- jednostki idiopatyczne
- zespoły paranowotworowe
- element składowy mieszanej choroby tkanki łącznej (MCTD- mixted connective tissue disease)
Miopatie zapalne jatrogenne
po leczeniu α-interferonem, przeszczepie szpiku kostnego, przeszczepie narządów wewnętrznych, w przebiegu „graft versus host disease”
Miopatie zapalne infekcyjne:
- Sarkoidoza (ziarniniakowe zapalenie mięśni)
- Włośnica
- Toksoplazmoza
- Schistosomiaza
- AIDS – HIV, infekcje oportunistyczne
- HTLV-1, inne retrowirusy
- Coxsackievirus
- Echowirus
Miopatie metaboliczne/endokrynologiczne:
- Nadczynność/ niedoczynność tarczycy
- Choroba/ zespół Cushinga
- Akromegalia
- Nadczynność przytarczyc
- Hiperaldosteronizm
- Niedoczynność przysadki możgowej
- Zespoły złego wchłaniania
- Jatrogenna miopatia sterydowa
Miopatie toksyczne:
- Alkohol (ostra miopatia nekrotyzująca, miopatia przewlekła)
- Kortykosterydy
- Leki blokujące transmisję n-m (pacjenci OIOM)
- Statyny, fibraty
- Zidovudyna i inne leki antyretrowirusowe
- D-penicylamina
110. Dystrofie mieśniowe
- dystrofia Duchenne’a
- Beckera
- twarzowo- łopatkowo-ramieniowa
- obręczowo-kończynowa
- Emery-Dreifussa
- ocznogardłowa
Zróżnicowane typy dziedziczenia, lokalizacje defektów genetycznych i produkty nieprawidłowych genów
Zróżnicowane obrazy kliniczne: wiek zachorowania, tempo progresji, rozkład zajętych mięśni, stopień niesprawności ruchowej, obecność/ nieobecność przerostów mięśniowych, przykurczów ścięgnowych, deformacji kręgosłupa i stawów, zajęcia mięśnia sercowego
111. Przełom miasteniczny i cholinergiczny- objawy i postępowanie
Przełom (kryza) - niewydolność oddechowa wymagająca intubacji i wspomaganej wentylacji, to nagłe nasilenie objawów miastenii połączone z wystąpieniem zaburzeń oddechowych
- Przełom miasteniczny – zaostrzenie choroby
- Przełom cholinergiczny – skutek przedawkowania leków antycholinesterazowych (zatrucie cholinergiczne muskarynowe i nikotynowe)
- U wielu chorych przełom mieszany - we włóknach mięśniowych blok depolaryzacyjny lub polaryzacyjny
Przyczyny:
- infekcje
- zaburzenia hormonalne np. nadczynność tarczycy
- poród, a zwłaszcza połóg
- narkoza z użyciem zwiotczających środków kurraropodobnych
- ciężki wysiłek fizyczny
- sytuacje stresowe
- przebywanie w wysokiej temperaturze
- leki blokujące synapsę nerwowo–mięśniową, np. penicylamina, aminoglikozydy, beta–blokery, niektóre leki psychotropowe, acetazolamid, lit, fenytoina, preparaty tarczycy
- zbyt mała dawka leków antycholinoesterazowych przyjmowanych przez
- chorego
- steroidoterapia w ciągu pierwszych kilku– kilkunastu dni
Objawy przełomu miastenicznego
- objawy wegetatywne:
- pocenie
- ślinotok
- tachykardia
- niepokój
- rozszerzenie źrenic
- cechy niewydolności oddechowej:
- duszność
- tachypnoe
- nasilenie męczliwości mięśni
- po podaniu i.v. 10 mg chlorku edrofonium następuje natychmiastowa poprawa trwająca kilka– kilkanaście minut
Przełom cholinergiczny występuje w przypadku przedawkowania leków antycholinesterazowych
Objawy przełomu cholinergicznego:
- występują objawy muskarynopodobne:
- biegunka
- zlewne poty
- nudności i wymioty
- bóle brzucha
- zaburzenia oddechowe
- objawy nikotynopodobne:
- nużliwość
- drżenia pęczkowe mięśni
- zaburzenia oddechowe
- objawy ośrodkowe:
- niepokój
- senność
- zab. przytomności
- źrenice zwężone
- ustępuje na krótko po podaniu leku antycholinergicznego
- po podaniu chlorku edrofonium krótkotrwałe pogorszenie
Postępowanie:
- OIOM – intubacja i sztuczna wentylacja (kilka/ kilkanaście dni)
- Identyfikacja i leczenie czynników odpowiedzialnych za przełom (infekcja dróg oddechowych lub inna, interakcje lekowe, przedawkowanie leków p/miastenicznych)
- Glikokortykosteroidy: korekta terapii immunosupresyjnej (↑ dawki prednisonu, wymiana osocza, IVIG, włączenie cytostatyku)
- Plazmafereza (5-6 zabiegów)
- IVIG - 400 mg/kg/d przez 5–6 dni
- Leki antycholinestrazowe – czasowe odstawienie konieczne w kryzie cholinergicznej, korzystne w każdym przełomie ; atropina i.v./i.m. znosi efekt muskarynowy przedawkowania
112. Miotonia, dystrofia miotoniczna
Miotonia to utrzymywanie sięskurczu mięśnia mimo próby jego rozkurczu.
Występuje w:
- dystrofii miotonicznej
- paramiotonii wrodzonej
- porażeniu okresowym hiperkaliemicznym
- miotonii wrodzonej
Miotonia:
- Utrudniona/ wydłużona relaksacja mięśnia po skurczu dowolnym
- Sztywność mięśni
- Niekiedy przerost mięśni
- Przemijające osłabienia/ niedowłady po wysiłku lub podczas wysiłku
113. Podziały neuropatii
114. Przyczyny polineuropatii
115. Polineuropatie – badania dodatkowe, leczenie, symptomatologia ogólna
116. Neuropatie obwodowe w cukrzycy
- dystalna, symetryczna polineuropatia zwykle czuciowa, czasem objawy ruchowe i autonomiczne
- proksymalna, asymetryczna neuropatia ruchowa, amiotrofia cukrzycowa, szybko występujące dolegliwości bólowe, zaniki mięśni
- neuropatia autonomiczna
- mononeuropatie z uwięźnięcia
- uszkodzenia nerwów czaszkowych, zwłaszcza III i VI
- wieloogniskowe zapalenie nerwów
117. Neuropatia autonomiczna w cukrzycy
- gastropareza cukrzycowa
- biegunki
- zaburzenia rytmu serca
- niedociśnienie ortostatyczne
- zaburzenia erekcji
118. Zespół Guillain-Barre: kryteria
Kryteria:
Objawy konieczne do rozpoznania GBS
- Postępujące osłabienie więcej niż jednej kończyny
- Zniesienie lub osłabienie odruchów
- Podwyższone stężenie białka w PMR przy niskiej cytozie
- Nieprawidłowości w EMG
- Stwierdzenie odcinkowej demielinizacji/remielinizacji w bioptacie nerwu
- Wykluczenie innych chorób: porfirii, nowotworu, choroby układowej, ołowicy.
Objawy przemawiające za rozpoznaniem GBS
- Narastanie objawów w okresie kilku dni- kilkunastu tygodni
- Względna symetryczność objawów
- Niewielkie nasilenie objawów czuciowych
- Zajęcie nerwów czaszkowych
- Początek zdrowienia 2-4 tygodnie od momentu zatrzymania postępu choroby
- Dysautonomia
- Brak gorączki w okresie początkowym choroby
- Podwyższone stężenie białka w PMR po tygodniu od początku choroby
- Nieprawidłowości w EMG.
119. GBS: postacie, obraz kliniczny
- klasyczna ruchowa polineuropatia zapalno-demielinizacyjna
- polineuropatia aksonalna (zespół AMAN, acute motor axonal neuropathy)
- zespół polineuropatii zapalno-demielinizacyjnych z zajęciem nerwów czuciowych (AMSN, acute motor sensory neuropathy
Zespół AMAN
- częsty zwłaszcza u dzieci
- przebiega szczególnie ciężko
- narastanie objawów trwa zwykle około 2 tygodni, po czym po krótkim plateau następuje pełny rozwój choroby
- polineuropatii towarzyszą liczne objawy ze strony przewodu pokarmowego
- ma się rozwijać w przebiegu zakażenia Campylobacter jejuni
- przeciwciała wytwarzane
w odpowiedzi na drobnoustrój wchodzą w krzyżową reakcję ze składnikami neurolemmy (gł. gangliozydami GM1 i GD1a) i w tej sposób wywołują objawy polineuropatii.
Zespół Millera-Fishera
- obok porażenia nerwów obwodowych występuje porażenie zewnętrznych nerwów gałkoruchowych i często towarzysząca ataksja.
- U wszystkich chorych z tym zespołem wykrywa się przeciwciała przeciw gangliozydowi GMQ1b, wobec
czego istotnym elementem diagnostyki jest serologia.
120. GBS: badania dodatkowe, leczenie
Postępowanie przyczynowe polega na stosowaniu plazmaferezy i dożylnych wlewów immunoglobulin (IVIG). U części pacjentów niezbędne jest postępowanie objawowe, polegające na leczeniu zaburzeń autonomicznych. U wszystkich chorych z niedowładami zaleca się fizjoterapię i rehabilitację ruchową kończyn.
121. Polineuropatie genetycznie uwarunkowane: krótka charakterystyka
Dziedziczne neuropatie ruchowo-czuciowe (HMN) - choroba Charcota-Marie-Tootha
- CMT typu I neuropatia demielinizacyjna z hipertrofią
- CMT typu II neuropatia aksonalna
- CMT typu III demielinizacyjna
- choroba Refsuma
- złożone postaci
Dziedziczne neuropatie czuciowe
Dziedziczne neuropatie ruchowe
Dziedziczne neuropatie czuciowe i autonomiczne (HSAN)
- rodzinna dysautonomia (zespół Rileya-Daya) - dziedziczenie AR
122. Najczęstsze mononeuropatie z ucisku:
Ucisk nerwu pośrodkowego
- zespół cieśni nadgarstka
- mrowienie, ból, drętwienie
- osłabienie mięśni kłębu kciuka
- zaburzenia czucia okolicy dłoniowej 31/2 palca od strony łokciowej (kciuka)
- [https://pracownia-emg.pl/test-phalena-i-tinela/] (opukiwanie nadgarstka powoduje ból i mrowienie)
- dodatni manewr Phalena
Ręka szponiasta: ucisk nerwu łokciowego
- zwykle po złamaniu kości łokciowej
- osłabienie i zanik mięśni
- osłabienie czucia 1 1/2 palca od strony łokciowej (mały palec i serdeczny)
Zespół sobotniej nocy
- ucisk nerwu promieniowego przez kość ramienną
- opadanie ręki w nadgarstku
- niedowład mięśnia ramienni-promieniowego i prostowników palców
- osłabienie czucia w okolicy tabakierki i po stronie grzbietowej ręki nad pierwszymi dwiema kośćmi śródręcza
stopa końsko-szpotawa
123. Zaburzenia węchu
Zaburzenia węchu:
- ilościowe
- jakościowe - wypaczone doznania lub iluzje węchowe:
- osłabienie lub zanik węchu (anosmia, hyposmia), nadmierne odczuwania węchu (hiperosmia)
- kakosmia
- kakogeusia
- omamy węchowe: napady padaczkowe skroniowe (“hakowe”), inne choroby płata skroniowego, zaburzenia psychotyczne, upośl. rozpoznawania doznań węchowych: agnozja węchowa
Anosmia (okresowa, stała)
- jednostronna (zwykle centralna, nieuświadomiona)
- dwustronna (zwykle obwodowa, uświadomiona)
Przyczyny
- guzy rynienki węchowej, płata czołowego, przysadki
- kostniak kości czołowej
- endokrynopatie: choroba Addisona, tyreotoksykoza
- tętniak tętnicy łączącej przedniej
- infekcje wirusowe, grypa, opryszczka - zniszczenie nerwów
- złamanie przedniego dołu czaszki
124. Przebieg analizatora wzrokowego i ubytki pola widzenia w zależności od lokalizacji uszkodzenia

Mroczek środkowy (uszkodzenie n. II)
- zapalenie pozagałkowe n. II
- ucisk nerwu II w kanale n. II (guzy, ziarniniaki, choroba Pageta)
- przyczyny śródczaszkowe: guzy, tętniaki
Ślepota jednooczna (uszkodzenie n. II)
- zapalenie nerwu II
- niedokrwienie, uraz
Kwadrantopsja dwuskroniowa/Niedowidzenie dwuskroniowe
- guzy przysadki
- raki nosogardła
- guzy III komory
Niedowidzenie dwunosowe
- tętniaki tętnic szyjnych wewnętrznych
- zlepne zapalenie pajęczynówki
Niedowidzenie połowicze
- pasmo wzrokowe z ubytkiem widzenia centralnego
- pasmo wzrokowe blisko skrzyżowania z zachowanym widzeniem centralnym
- pasmo wzrokowe blisko promienistości wzrokowej
Kwadrantopsja - uszkodzona promienistość wzrokowa
- dolna - uszkodzenie części ciemieniowej
- górna - uszkodzenie części skroniowej promienistości
Ślepota korowa
- Nagłe obustronne osłabienie lub utrata wzroku
- Brak reakcji na nastawność, prawidłowa reakcja na światło
- Brak odruchu mrugnięcia na ruch przedmiotu
- Prawidłowe ruchy gałek ocznych
- Brak oczopląsu optokinetycznego
125. Uszkodzenie nerwów gałkoruchowych
Uszkodzenie nerwu III:

- Podwójne widzenie przy spojrzeniu we wszystkich kierunkach
- Rozbiezne ustawienie gałki ocznej
- Ptoza
- Poszerzenie źrenicy ze zniesieniem reakcji na swiatło i nastawienie
Uszkodzenie nerwu IV:

- Pionowa i skośna diplopia przy patrzeniu w dół i przyśrodkowo
- Łagodna rotacja gałki ocznej na zewnątrz i ku górze
- Pacjent przechyla głowę w stronę przeciwną do porażenia
Przyczyny:
- demielinizacja
- udar niedokrwienny
- glejak
- przerzuty
Uszkodzenie nerwu VI:

- Pozioma diplopia, największa przy patrzeniu w bok po stronie uszkodzenia
Przyczyny:
- zespół Gradeniga
- zakrzep zatoki jamistej
- złamanie lub naciek nowotorowy podstawy czaszki
- zespół szczeliny oczodołowej górnej
126. Przyczyny uszkodzenia nerwu III
Przyczyny:
uszkodzenie pnia:
- tetniak tętnicy łączącej tylnej
- zakrzepica zatoki jamistej
- patologia szczeliny oczodołowej górnej (naciek nowotworowy)
- cukrzyca
uszkodzenie jądra w śródmózgowiu:
- udar niedokrwienny
- SM
- glejak
- przerzuty nowotworowe
127. Zespół zatoki jamistej i szczeliny oczodołowej górnej
Zatoka jamista (uszkodzenie nerwów III, IV, VI, V1)
- guzy (oponiaki, przerzuty, guzy przysadki)
- tętniak wewnątrzjamisty
- zakrzep zatoki jamistej
Oczodół (uszkodzenia jak zatoka jamista)
- guzy oczodołu
- ziarniniaki
- zapalenie okostnej lub kości
Nerw VI ze względu na długi przebieg wewnątrzczaszkowy jest szczególnie “uraźliwy”
128. Skojarzone ruchy gałek ocznych i przyczyny ich zaburzeń
Ruchy śledzenia: precyzyjne, płynne, zwykle mimowolne podążanie za przedmiotem z fiksacją wzroku i utrzymanie ostrego obrazu w jednym miejscu plamek
Ruchy spojrzenia (sakkadowe): nagłe, szybkie, zwykle nieuświadomione ruchy skojarzone gałek, pozwalające na nagłą zmianę obserwowanego obiektu, bez utrzymywania ostrego obrazu w jednym miejscu plamek (upośledzone w chorobie Parkinsona, PPN)
Napady padaczkowe (czołowe):zrywające zbaczanie w stronę przeciwną do ogniska czołowego, zgodne do zajętych kończyn
Krwotok mózgowy (rozległy udar niedokrwienny) z zajęciem wzgórza: stałe zbaczanie gałek ocznych w stronę ogniska, przeciwną do porażonych kończyn (chory patrzy na ognisko)
Krwotok do pnia (most - przyśrodkowa część tworu siatkowatego): stałe zbaczanie gałek w stronę przeciwną do ogniska, zgodną z porażeniem (chory patrzy na porażenie)
- upośledzenie ruchomości gałek ocznych ku górze i ruchu zbieżności, porażenie nerwu III i IV
- reakcja źrenic na światło i zbieżność upośledzona
- źrenice rozszerzone
Przyczyny
- guzy III komory i szyszynki
- wodogłowie
- stwardnienie rozsiane
- encefalopatia Wernickego
- zapalenie mózgu
Porażenie międzyjądrowe
Uszkodzenie pęczka podłużnego przyśrodkowego
Przyczyny
- stwardnienie rozsiane
- zawały i krwotoki pnia mózgu
- urazy pnia mózgu
- jamistość opuszki
- zatrucie fenytoiną
129. Zaburzenia ruchomości źrenic
Rozszerzenie źrenicy (mydriasis)
- uszkodzenie nerwu III
- napad migreny
- leki antycholinergiczne (atropina)
- leki antyhistaminowe
- trójcykliczne przeciwdepresyjne
- doustne antykoncepcyjne
Zwężenie źrenicy (miosis)
- zespół Hornera
- krwotok do mostu
- leki: opioidy, cholinolityki, mirtazapina
- zwężenie źrenicy
- zwężenie szpary powiekowej
- upośledzenie potliwości na twarzy (uszkodzenie poniżej rozwidlenia tętnicy szyjnej)
Przyczyny
- rdzeń szyjny: guzy, jamistość rdzenia
- korzenie rdzeniowe C8/Th1, nerwiaki
- dolna część splotu ramiennego
- tętnica szyjna wewnętrzna (rozwarstwienie)
- część szyjna pnia współczulnego (zespół Pancoasta)
- brak reakcji na światło
- prawidłowa reakcja na zbieżność i nastawność
- nierówność źrenic
Przyczyny
- kiła układu nerwowego
- uszkodzenie śródmózgowia
- choroby naczyniowe, guzy, zapalenie, SM
- neuropatia cukrzycowa i alkoholowa
Zespołu Argylla-Roberstona nie należy mylić z podobnymi objawami u ludzi w podeszłym wieku.
130. Symptomatologia kliniczna i przyczyny uszkodzenia nerwu V
Przyczyny uszkodzenia nerwu V
Oczodół, szczelina oczodołowa górna, zatoka jamista (V1, III, IV, VI)
- zmiany zapalne, nowotwory, tętniak
Kości czaszki
- nowotwory nosogardła, przerzutowe
- urazy oczodołu
Kąt mostowo-móżdżkowy
- nowotwory
- podostre lub przewlekłe zapalenie opon
Zwój Gassera
- neuralgia n V
Most
- choroby naczyniowe
- nowotwory
- SM
- jamistość opuszki
Inne przyczyny (uszkodzenie o różnej lokalizacji)
- cukrzyca
- toczeń rumieniowaty układowy
131. Niedowład twarzy a porażenie nerwu VII. Symptomatologia kliniczna i przyczyny uszkodzenia nerwu VII
Przebieg nerwu VII:
- nerw VII utworzony jest z włókien ruchowych, zaopatrujących mięśnie mimiczne twarzy, oraz z nerwu pośredniego,prowadzącego włókna smakowe z przednich 2/3 języka i włókna przywspółczulne dla gruczołów ślinowych i mięśnia strzemiączkowego
- jądro ruchowe znajduje się w bocznej części mostu, przed opuszczeniem mostu w kącie mostowo-móżdżkowym włókna nerwu tworzą pętlę zawijającą się wokół jądra VI nerwu
- nerw wychodzi w kącie mostowo-móżdżkowym i przechodzi do przewodu słuchowego wewnętrznego razem z nerwem pośrednim i VIII nerwem czaszkowym
- wnika w głąb kości skalistej do ucha środkowego, nerw pośredni do zwoju kolanka
- nerw skalisty większy opuszcza zwój kolanka prowadząc włókna przywspółczulne dla gruczołu łzowego
- nerw VII biegnie w kanale nerwu twarzowego w części skalistej kości skroniowej, oddaje gałązkę do mięśnia strzemiączkowego, regulującą wrażliwość kosteczek słuchowych na dźwięk
- oddaje strunę bębenkową, zaopatrującą smakowo 2/3 języka i prowadzącą włókna przywspółczulne dla podżuchwowych i podjęzykowych gruczołów ślinowych
- pień nerwu VII wychodzi otworem rylcoowo-sutkowym wnika do ślinianki przyusznej i dzieli się na gałęzie zaopatrujące mięśnie mimiczne twazry
Niedowład ośrodkowy jednostronny
- spowodowany nadjądrowym uszkodzeniem GNR po stronie przeciwnej
- przyczyny: udar niedokrwienny, guz
Niedowład jednostronny obwodowy
- uszkodzenie jądra VII, zwoju kolanka lub pnia nerwu VII
- na poziomie: mostu, kąta mostowo-móżdżkowego, w kanale nerwu, zwoju kolanka, obwodowe gałęzie nerwu
Obustronny niedowład obwodowy
- obustronne porażenie Bella, borelioza, sarkoidoza, GBS, miastenia, miopatie
132. Symptomatologia kliniczna i przyczyny uszkodzenia nerwu VIII
Przyczyny:
- zespół kąta mostowo-móżdżkowego
- złamanie piramidy kości skroniowej
- zapalenie nerwu przedsionkowego (neuronitis vestibularis)
Objawy:
- zawroty głowy
- oczopląs poziomy lub obrotowy
- faza szybka oczopląsu przeciwna do uszkodzenia
- pacjent chodząc zbacza w stronę uszkodzenia
- głuchota typu odbiorczego
- szumy uszne
- ewent. zajęte nerwy V i VII
133. Zapalenie naczyń, etiopatogeneza, obraz kliniczny
- Patologia: obecność immunologicznych
- kompleksów w ścianach naczyń z zapaleniem
- pod postacią wielojądrzastych leukocytów,
- eozynofili, z martwicą ściany.
- Zajęcie układu nerwowego / ok. 25%/
- Polineuropatia czuciowo-ruchowa
- Mononeuritis multiplex
- Udary / niedokrwienne, krwotoczne/
- Drgawki
- Bóle i zawroty głowy
- Ubytki widzenia jednooczne
- Psychozy
134. Uszkodzenie układu nerwowego w zespole antyfosfolipidowym
Zespół antyfosfolipidowy – kryteria:
Kryteria kliniczne:
- trzy lub więcej poronienia samoistne przed 10. tygodniem ciąży
- jedna lub więcej niewyjaśniona śmierć płodu po 10. tygodniu ciąży
- ciężki stan przedrzucawkowy lub niewydolność łożyska - poród przed 34. tygodniem ciąży
- niewyjaśniona zakrzepica żylna bądź tętnicza
- zakrzepica drobnych naczyń, bez cech zapalenia
Kryteria laboratoryjne:
- średnie lub wysokie miano przeciwciał antykardiolipinowych klasy IgM lub IgG w 2 niezależnych pomiarach w odstępie minimum 6-tygodniowym
- obecność antykoagulantu tocznia w surowicy w 2 niezależnych pomiarach w odstępie minimum 6 tygodniowym
Zespół antyfosfolipidowy rozpoznajemy gdy jest spełnione przynajmniej jedno kryterium kliniczne oraz jedno kryterium laboratoryjne.
Jeżeli zmiany występują w więcej niż 3 narządach mówimy o ciężkim zespole antyfosfolipidowym.
Objawy:
- zmiany zakrzepowo-zatorowe
- powikłania ginekologiczne
Zaburzenia neurologiczne w APS:
- Ogniskowe niedokrwienie mózgu, TIA
- Zakrzepy naczyń żylnych
- Otępienie wielozawałowe
- Powkłania oczne (ślepota korowa, ostra neuropatia oczna)
- Zespół SM-like
- Poprzeczne uszkodzenie rdzenia
- Migrena i migreno-podobne bóle głowy)
- Zaburzenia psychiczne
135. Zaburzenia neurologiczne w niedoborach witaminowych
- B1- polineuropatia, choroba beri-beri, encefalopatia Wernickiego, zespół Korsakowa
- B6 - polineuropatia czuciowa, napady padaczkowe
- B12 – niedokrwistość Adisona-Biermera, zwyrodnienie tylnosznurowe rdzenia, zaburzenia psychiczne, otępienie, niedowład spastyczny kończyn dolnych, zaburzenia czucia proprioceptywnego, parestezje
- E - neuropatia czuciowa grubych włókien, zwyrodnienie rdzenia kręgowego i móżdżku
- D - miopatia proksymalna
136. Encefalopatia wątrobowa, patogeneza, obraz kliniczny
- Przyczyna: wzrost amoniaku w surowicy krwi, np. po krwawieniu do przewodu pokarmowego, w infekcji albo przedawkowaniu diuretyków
- Ostra lub wrotna: (szybki przebieg; drgawki, delirium)
- Przewlekła: zaburzenia psychiczne, ataksja, dyzartria, parapareza spastyczna, zaburzenia pozapiramidowe, móżdżkowe, zaburzenia świadomości
Patogeneza:
- neurotoksyny: amoniak, merkaptany, kwasy tłuszczowe, fenole
- fałszywe neuroprzekaźniki: β-fenyloetanoloamina, oktopamina
Objawy:
- zaburzenia cyklu snu i czuwania
- zaburzenia uczenia się, zapamiętywania, koncentracji
- zaburzenia orientacji przestrzennej
- zaburzenia nastroju - apatia, nadpobudliwość
- asterixis (grubofaliste drżenie rąk)
- foetor hepaticus
- zmiany w zapisie EEG - początkowo zwolnienie czynności podstawowej, następnie ogniskowe lub uogólnione fale wolne, fale trójfazowe, wyładowania napadowe iglic lub fal wolnych
- zwiększone stężenie amoniaku we krwi
- w ostatnim etapie śpiączka wątrobowa, głębokie zaburzenia metaboliczne (kwasica metaboliczna)
Pieciostopniowa klasyfikacja 0-5
Testy psychometryczne do oceny 0-2
- test łączenia punktów
- test labiryntu
- test gwiazdy
Leczenie:
- dieta ubogobiałkowa
- neomycyna albo metronidazol
- asparaginian ornityny
137. Powikłania neurologiczne w nadczynności i niedoczynności tarczycy
Nadczynność tarczycy
- Proksymalna miopatia: (osłabienie i zanik mm.dosiebnych, fascykulacje)
- Zaburzenia oczne: (wytrzeszcz, podwójne widzenie z ograniczeniem ruchów gałek do góry), opadanie powiek jednostronne
- Drżenie
- Rzadko: drgawki uogólnione, pląsawica, neuropatia obwodowa, uszkodzenie dróg piramidowych
- Badania dodatkowe: emg, umiarkowana nadczynność tarczycy, leczenie przyczynowe
Niedoczynność tarczycy
- Otępienie
- Bóle głowy
- Zaburzenia psychiczne: (psychozy, urojenia, halucynacje)
- Ataksja móżdżkowa
- Neuropatia z „uwięźnięcia”,
- Bóle, kurcze i przerost mięśni
138. Najważniejsze zespoły neurologiczne w przewlekłym alkoholizmie
- Padaczka
- Neuropatia alkoholowa
- Zwyrodnienie i zanik móżdżku
- Otępienie alkoholowe
- Udary mózgu
- Miopatia
- Choroba Marchiafavy-Bignamiego
- Encefalopatia Wernickiego
- Zespół Korsakowa
139. Zespoły paranowotworowe
Zespół paraneoplastyczny: Jest to zespół objawów kliniczno–neuropatologicznych występujący w przebiegu choroby nowotworowej spowodowany jej „ pośrednim” (w sensie czasu i lokalizacji) działaniem na układ nerwowy.
Neurologiczne zespoły paranowotworowe
Rdzeń kręgowy:
- Podostra nekrotyzujaca mielopatia
- Choroba neuronu ruchowego (4-10%)
Nerwy obwodowe i korzenie:
- Podostra neuropatia czuciowa.
- Czuciowo-ruchowa neuropatia obwodowa.
- Ostra poliradikuloneuropatia.
- Ostre zapalenie splotu barkowego.
- Neuropatie z niedożywienia.
Połączenie nerwowo-mięśniuowe i mięśnie:
- Neuromiopatia z wyniszczenia
- Neuromiopatia rakowa
- Ostra neuromiopatia
- Zapalenie mięśniowe i skórno-mięśniowe
- Zespół miasteniczny (LEMS)
Mózg i nerwy czaszkowe:
- Demencja
- Zapalenie mózgu limbiczne
- Podostre rakopochodne zwyrodnienie mózdzku (2%)
- Wczesnodziecięca encefalopatia miokloniczna
- Zapalenie nerwów wzrokowych
Klasyfikowane w zależności od rodzaju przeciwciała
- Anty-Ho
- Anty-Yo
- Anty-Ri
- Anty-CV2
Mogą współwystępować dwa rodzaje przeciwciał CANA oraz inne przeciwciała antyneuronalne
Często współistnieją zespoły kliniczne zajęcia OUN, ObUN, złącza nerwowo-mięśniowego
Podłoże autoimmunologiczne
Przeciwciała p/onkoneuronalnym proteinom (CANA-circulating anti-neuronal antibodies)
Przypadki seropozytywne i seronegatywne (przyczyna czy następowe uszkodzenie tkanki)
Udział mechanizmów komórkowych (cytotoksyczne limfocyty T, aktywacja makrofagów)
Zespoły anty-Ho
- SSN (subacute sensory neuropathy)
- podostra neuropatia czuciowa
- Encephalomyelitis
- Limbic encephalitis
- Brain steam encephalitis
- Myelitis
- Ganglioradiculoneuritis
- Autonomic neuropathy
- Najczęściej w przebiegu raka drobnokomórkowego płuc
- Czułość przeciwciał 30-40%
Zespoły anty-CV2
- Zwyrodnienie móżdżku
- Zapalenie tęczówki/neuropatia nerwu wzrokowego
- Polineuropatia czuciowo-ruchowa /typ aksonalno-demielinizacyjny/
- LEMS
- Przeciwciała anty-CV2 reagują z antygenem nerwu obwodowego
- W 20% przypadków współistnieją przeciwciała anty-Ho
Leczenie choroby podstawowej
Immunoterapia - ogólna kontrowersje
- Plazmafereza
- Kortykosterydy
- cyklofosfamid
140. Zaburzenia neurologiczne w chorobach układowych tkanki łącznej
- bolesne mononeuropatie, w SLE może rozwinąć się symetryczna neuropatia czuciowo-ruchowa, neuropatia z uwięźnięcia w RZS, neuropatia nerwu trójdzielnego w zespole Sjogrena
- zaburzenia neuropsychiatryczne - napady drgawek lub psychoza - są jednym z kryteriów SLE
- w zespole antyfosfolipidowym zakrzepica i udary mózgu, stan przedrzucawkowy, migreny
W guzkowym zapaleniu tętnic
- 25% objawy neurologiczne
- mononeuropatia wieloogniskowa (najczęściej porażenie nerwu strzałkowego)
- udary mózgu
- polineuropatia czuciowo-ruchowa
- mononeuritis multiplex
- drgawki, bóle i zawroty głowy
- ubytki widzenia jednooczne
- psychozy
W RZS:
- zespół cieśni nadgarstka
- neuropatia wieloogniskowa
- zaniki mięśni
- radikulopatia szyjna na wysokim poziomie (podwichnięcie szczytowo-obrotowe)
- zespół miasteniczny u pacjentów leczonych penicylaminą
- miopatia posterydowa
W SLE:
- (cerebral lupus) padaczka
- otępienie
- zaburzenia psychopatologiczne
- neuropatia wieloogniskowa
- pląsawica
- uszkodzenia nerwów czaszkowych
- zapalenie poprzeczne rdzenia
- polineuropatia czuciowo-ruchowa
- zapalenie nerwowo-mięśniowe
- bóle głowy
- wariant naśladujący SM (SM-like) z demielinizacją
- ubytki w polu widzenia.
W APS:
- Ogniskowe niedokrwienie mózgu, TIA
- Zakrzepy naczyń żylnych
- Otępienie wielozawałowe
- Powkłania oczne (ślepota korowa, ostra neuropatia oczna)
- Zespół SM-like
- Poprzeczne uszkodzenie rdzenia
- Migrena i migrenopodobne bóle głowy
- Zaburzenia psychiczne
W zespole Sjogrena:
- neuropatia obwodowa, głównie czuciowa
- zespoły z uwięźnięcia
- mielopatia
- miopatia proksymalna
- zapalenie opon i mózgu
- izolowana neuropatia nerwu V
141. Uszkodzenie układu nerwowego w białaczkach
Białaczki: porażenie nerwów czaszkowych, padaczka, niedowłady, afazja, polineuropatia, uszkodzenie rdzenia kręgowego, krwawienia śródczaszkowe, zaburzenia świadomości spowodowane leukostazą
Szpiczak mnogi: porażenie mięśni gałkoruchowych, niedowłady kończyn dolnych, polineuropatia, objawy ogniskowe, bóle głowy. W gammapatiach monoklonalnych rozwija się neuropatia przypominająca CIDP
Czerwienica: zaburzenia krążenia mózgowego
Makroglobulinemia Waldrenströma: zespół Binga-Neela, neuropatia czuciowo-ruchowa, udary mózgu, zespół rdzeniowy, zespół nadlepkości: bóle, zawroty glowy, senność, zab. wzrokowe, słuchowe, śpiączka
Chłoniaki złośliwe: zajęcie opon, nerwów czaszkowych, objawy guza mózgu, rdzenia
Ziarnica złośliwa: objawy rdzeniowe, mózgowe, zajęcia nn. obwodowych, splotu barkowego.
142. Zaburzenia neurologiczne w AIDS
Pierwotne ostre
- Zapalenie lub ostra encefalopatia
- Surowicze zapalenie opon
- Zapalenie rdzenia kręgowego
- Uszkodzenie pojedynczych nerwów czaszkowych
Pierwotne przewlekłe
- Otępienie podkorowe (ok.70%)
- Zapalenie opon z zajęciem n. czaszkowych V,VII,VIII
- Uszkodzenie rdzenia kręgowego (zwyrodnienie powrózkowe)
- Polineuropatia
Zakażenie wtórne oportunistyczne
- wirusowe (wirus herpes, cytomegalia, półpasiec, encefalopatia wieloogniskowa)
- grzybicze (cryptoccoccus neoformans, candida albicans)
- pierwotniaki (toxoplasma gondi)
- bakteryjne (kiła, grużlica, mycobacterium)
Nowotwory: (mięsak Kaposiego, chłoniak)
Udary mózgu: (udary krwotoczne, niedokrwienne)
HIV-leukoencefalopatia
miopatie związane z HIV
HIV-associated dementia complex
Herpesowe zapalenie mózgu
143. Zaburzenia neurologiczne w mocznicy
Niewydolność nerek:
- Encefalopatia mocznicowa, z zaburzeniami percepcji i halucynacjami wzrokowymi, prowadzi do śpiączki, występują asteriksje
- Polineuropatia mocznicowa
- odwracalny niedowład połowiczy w przebiegu dializy
- sztywność mięśni osiowych
- napady drgawkowe
- Zespół podializacyjny
- Encefalopatia dializacyjna
- postępująca neuropatia czuciowo-ruchowa, zwykle odpowiadająca na przeszczep (obwodowa neuropatia aksonalna, głównie czuciowa)
Powikłania po transplantacji nerek
- chłoniak mózgu (5% chorych po transplantacji)
- amyloidoza
- infekcje grzybicze oun
- objawy neurologiczne po cyklosporynie (napady padaczkowe, encefalopatia, neuropatia, miopatia)
- encefalopatia Wernickego
- postępująca wieloogniskowa encefalopatia (PML)
144. Przyczyny radikulopatii szyjnej
Przyczyny radikulopatii szyjnej
Nowotwory kanału kręgowego zewnątrzrdzeniowe
podtwardówkowe:nerwiaki, oponiaki
nadtwardówkowe: przerzuty, szpiczaki, mięsaki
Spondyloza szyjna
- zmiany zwyrodnieniowo-zniekształcające
- dyskopatie
Wady rozwojowe szyjnego odcinka kręgosłupa
Urazy
Zmiany zapalne
- gruźlica
- sarkoidoza
- zapalenie tętnic
145. Zespoły korzeniowe odcinka szyjnego kręgosłupa
Korzeń C6 (przestrzeń C5/C6)
- ból, parestezje i niedoczulica (boczna powierzchnia ramienia,promieniowa powierzchniaprzedramienia, kciuk i palec wskazujący)
- osłabienie odruchu z mięśnia dwugłowego
Korzeń C7 (przestrzeń C6/C7)
- ból, parestezje i niedoczulica (okolica międzyłopatkowa, tylna powierzchnia ramienia, łokciowa powierzchnia przedramienia, palec II i III )
- osłabienie odruchu z mięśnia trójgłowego
Korzeń C8 (przestrzeń C7/Th1)
- ból, parestezje i niedoczulica (tylna powierzchnia ramienia, łokciowa powierzchnia przedramienia, palec IV i V)
- osłabienie odruchu z mięśnia trójgłowego
- objaw Hornera
146. Mielopatia szyjna
- ból i sztywność karku
- bóle i parestezje obręczy barkowej
- zespół amiotroficzny w kończynach górnych (zaniki mięśni i odruchów głębokich, niedowłady)
- niedowład spastyczny w kończynach dolnych
- zaburzenia chodu
147. Zespół Pancoasta
Zespół Pancoasta (dolna część splotu)
- narastające bóle barku, głównie nocne parestezje i niedoczulica (tylna powierzchnia ramienia, łokciowa powierzchnia przedramienia, palec IV i V)
- brak odruchu z mięśnia trójgłowego
- osłabione: wyprost w stawie łokciowym, zginanie dłoniowe lub grzbietowe nadgarstka, zginanie i prostowanie palców
- objaw Hornera
- spowodowany uszkodzeniem dolnej części splotu C8-Th1
148. Ostry zespół bólowy kręgosłupa
Typ I
- tępy, rozlany, stopniowo narastający
- zaostrzenie podczas stania, ruchów tułowia, siedzenia
- leżenie łagodzi ból
Typ II (postrzał, lumbago)
- nagły, ostry, ból nasilany przez ruch
- lordoskolioza
- leżenie znosi ból
149. Zespoły korzeniowe odcinka lędźwiowo-krzyżowego
Korzeń S1
- ból , parestezje i niedoczulica „korzeniowa”
- dodatni objaw Laseque’a, Bragaarda
- osłabienie odruchu skokowego i podeszwowego
- osłabienie zginania podeszwowego stopyi palucha
- zanik mięśnia trójgłowego
Korzeń L5
- ból , parestezje i niedoczulica „korzeniowa”
- dodatni objaw Laseque’a, Bragaarda
- osłabienie zginania grzbietowego stopyi palucha
- zanik mięśnia piszczelowego przedniego
- prawidłowe odruchy głębokie
Korzeń L4
- ból , parestezje i niedoczulica „korzeniowa”
- dodatni objaw Mackiewicza
- osłabienie odruchu kolanowego
- osłabienie prostowania w stawie kolanowym
- zanik mięśnia czworogłowego
150. Zespół ogona końskiego
Zespół ogona końskiego:
- bóle krocza
- niedoczulica typu spodenek jeździeckich
- osłabienie odruchów skokowych, rzadziej kolanowych
- osłabienie zginania podeszwowego i/lub grzbietowego stóp i paluchów
- zaburzenia zwieraczy (inkontynencja moczu i stolca)
- asymetria objawów
Zespół stożka rdzeniowego – zespół neurologiczny spowodowany patologią końcowej części rdzenia kręgowego zwanej stożkiem (conus medullaris). Na obraz kliniczny zespołu składają się zaburzenia czucia w obszarze skóry okolicy perianalnej, perigenitalnej i wewnętrznej powierzchni ud, zaburzenia mikcji i funkcji seksualnych, oraz ból zajętej okolicy. Etiologia jest najczęściej urazowa, niekiedy naczyniowa (tętniak aorty) lub związana z guzem nowotworowym samego stożka (np. wyściółczakiem).
Zespół ogona końskiego – charakterystyczny zespół objawów neurologicznych, powstających wskutek uszkodzenia końcowego odcinka rdzenia kręgowego noszącego nazwę właśnie ogona końskiego.
Wskutek dyskopatii lub wzrostu guza w świetle końcowego odcinka kanału kręgowego dochodzi do ucisku na nerwy tworzące koński ogon, co powoduje poniższe objawy:
- silne bóle okolicy krocza i okolicy krzyżowej
- "spodenkowe" zaburzenia czucia
- porażenie zwieraczy (inkontynencja moczu i stolca)
- niedowład lub porażenie nerwu strzałkowego wspólnego
- brak odruchu ze ścięgna Achillesa.
Powyższe dwa objawy występują asymetrycznie, czyli tylko w jednej kończynie dolnej.
151. Diagnostyka zespołów bólowych kręgosłupa
- Badanie specjalistyczne
- neurolog
- ortopeda
- reumatolog
- diagnostyka patologii w miednicy mniejszej
- Badania radiologiczne (rtg, usg, MRI)
- Badanie elektrofizjologiczne
- Badania laboratoryjne
- Badanie płynu mózgowo-rdzeniowego
- Diagnostyka bólów rzutowanych
152. Leczenie zespołów bólowych kręgosłupa
- bezwzględne leżenie
- niesteroidowe leki p/zapalne
- leki obniżające napięcie mięśniowe
- łagodne leki uspokajające i p/depresyjne
- fizykoterapia
- Sollux, DDM, DKF, jontoforeza
- parafina
- kinezyterapia czynna



